
Late Pleistocene Altitudinal Segregation and Demography Define Future Climate Change Distribution of the Peromyscus mexicanus Species Group: Conservation Implications

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site, Nuclear DNA Extraction and Sequencing
2.2. Phylogeographic Patterns, Divergence Times and Demography
2.3. Current and Future Species Distribution Models
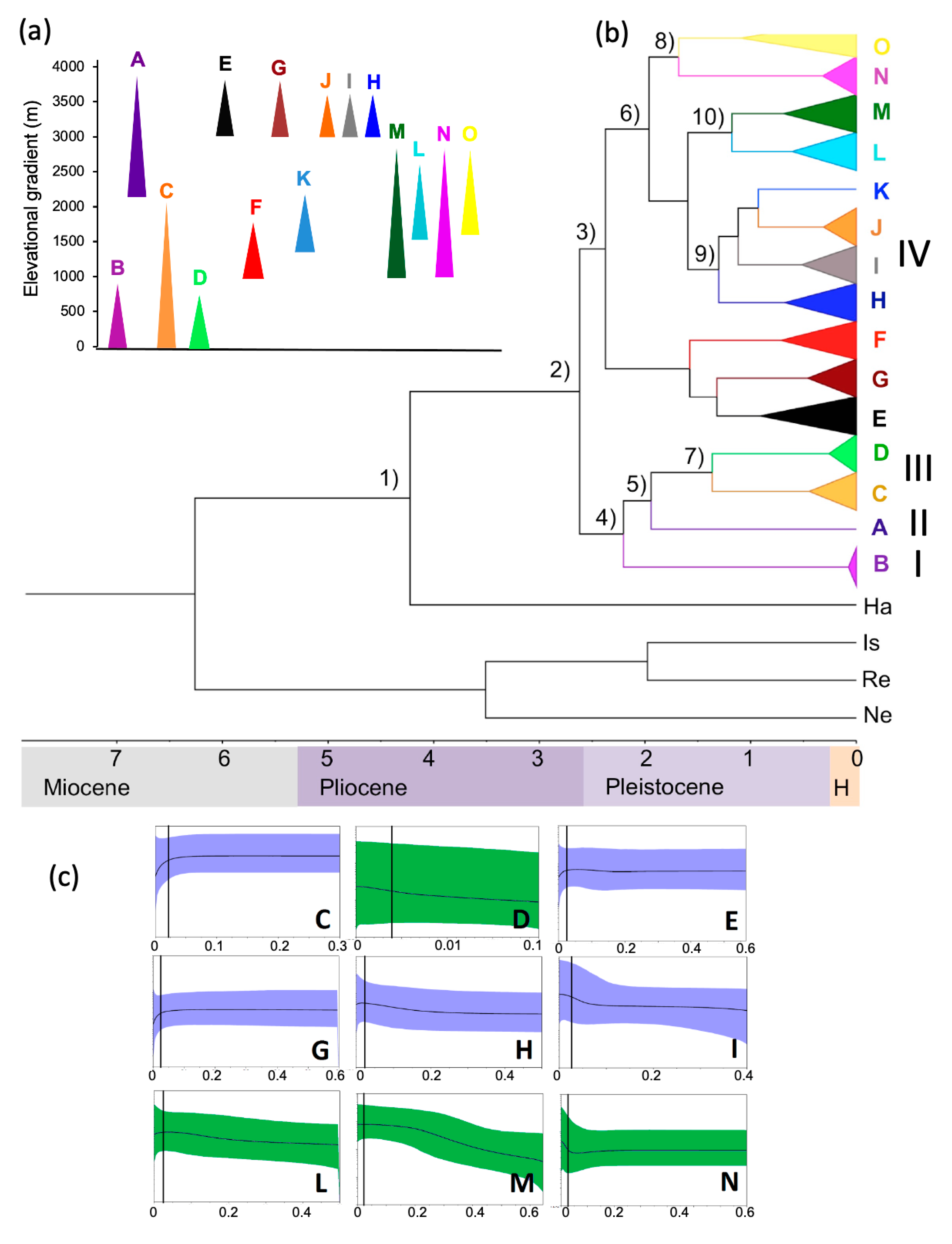
3. Results
3.1. Phylogeography and Demographic Patterns
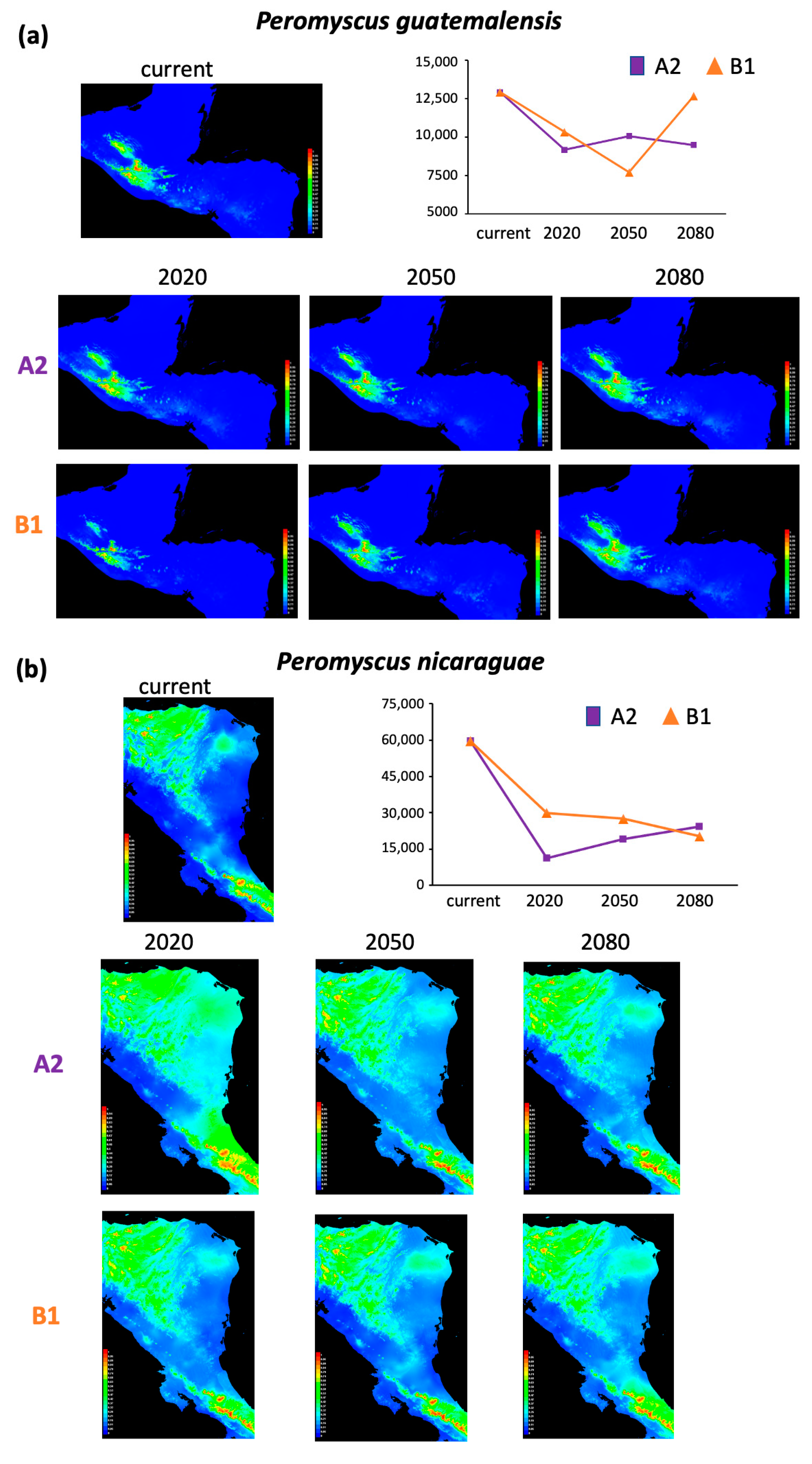
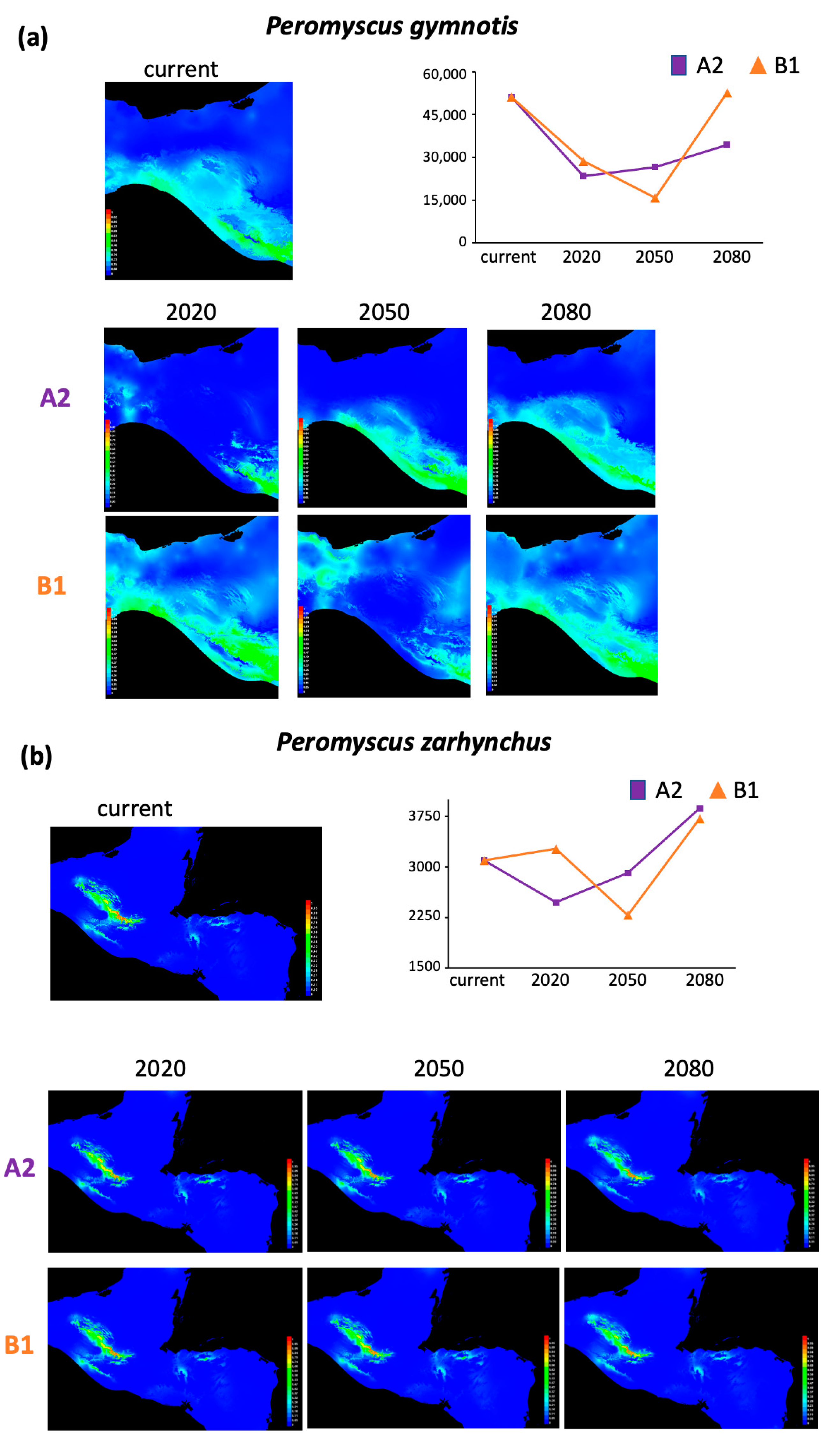
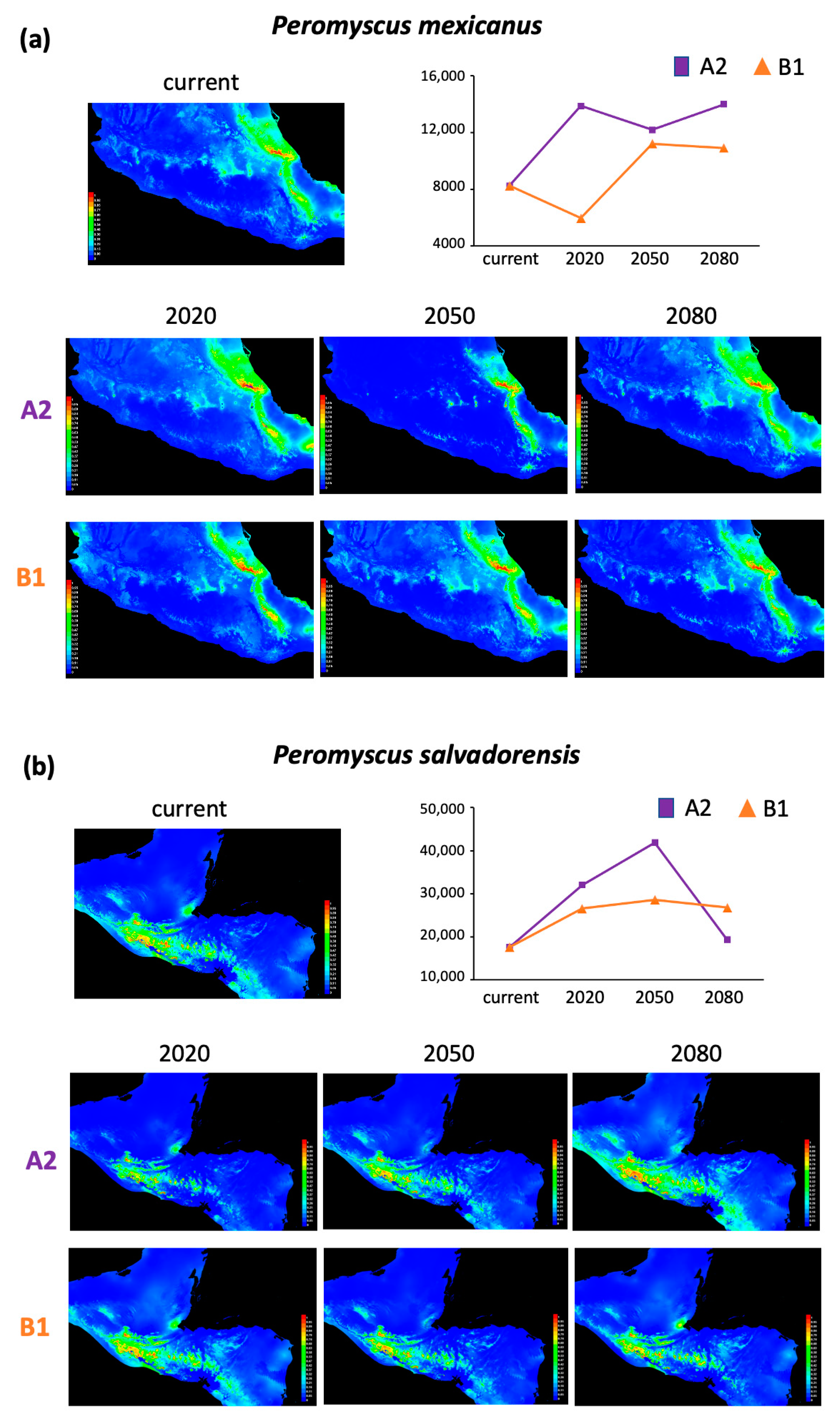
3.2. Niche Modeling
4. Discussion
4.1. Phylogeographic and Demographic Patterns of Lowland and Highland Mountain Lineages
4.2. Overall Distribution Range Changes Associated with Future Climate Change
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spehn, E.M.; Rudmann-Maurer, K.; Körner, C. Mountain biodiversity. Plant Ecol. Divers. 2011, 4, 301–302. [Google Scholar] [CrossRef]
- Fjeldså, J.; Bowie, R.C.K.; Rahbek, C. The role of mountain ranges in the diversification of birds. An. Rev. Ecol. Evol. Syst. 2012, 43, 249–265. [Google Scholar] [CrossRef]
- Antonelli, A.; Kissling, W.D.; Flantua, S.G.A.; Bermúdez, M.A.; Mulch, A.; Muellner-Riehl, A.N.; Kreft, H.; Linder, H.P.; Badgley, C.; Fjeldsa, J.; et al. Geological and climatic influences on mountain biodiversity. Nat. Geosci. 2018, 11, 718–725. [Google Scholar] [CrossRef]
- Kuntner, M.; Năpăruş, M.; Li, D.; Coddington, J.A. Phylogeny predicts future habitat shifts due to climate change. PLoS ONE 2014, 9, e98907. [Google Scholar] [CrossRef] [PubMed]
- Guevara, L.; Gerstner, B.E.; Kass, J.M.; Anderson, R.P. Toward ecologically realistic predictions of species distributions: A cross-time example from tropical montane cloud forests. Glob. Chang. Biol. 2018, 24, 1511–1522. [Google Scholar] [CrossRef]
- Kozak, K.H.; Wiens, J.J. Climatic zonation drives latitudinal variation in speciation mechanisms. Proc. R. Soc. B 2007, 274, 2995–3003. [Google Scholar] [CrossRef]
- Drovetski, S.V.; Semenov, G.; Drovetskaya, S.S.; Fadeev, I.V.; Redkin, Y.A.; Voelker, G. Geographic mode of speciation in a mountain specialist Avian family endemic to the Palearctic. Ecol. Evol. 2013, 3, 1518–1528. [Google Scholar] [CrossRef]
- Mastretta-Yanes, A.; Moreno-Letelier, A.; Piñero, D.; Jorgensen, T.; Emerson, B. Biodiversity in the Mexican highlands and the interaction of geology, geography and climate within the Trans-Mexican Volcanic Belt. J. Biogeogr. 2015, 42, 1586–1600. [Google Scholar] [CrossRef]
- Borja-Martínez, G.; Tapia-Flores, D.; Shafer, A.B.A.; Vázquez-Domínguez, E. Highland forest’s environmental complexity drives landscape genomics and connectivity of the rodent Peromyscus melanotis. Landsc. Ecol. 2022, 37, 1653–1671. [Google Scholar] [CrossRef]
- Hua, X.; Wiens, J.J. How does climate influence speciation? Am. Nat. 2013, 182, 1–12. [Google Scholar] [CrossRef]
- Bastianelli, G.; Wintle, B.A.; Martin, E.H.; Seoane, J.; Laiolo, P. Species partitioning in a temperate mountain chain: Segregation by habitat vs. interspecific competition. Ecol. Evol. 2017, 7, 2685–2696. [Google Scholar] [CrossRef] [PubMed]
- Cadena, C.D.; Kozak, K.H.; Gómez, J.P.; Parra, J.L.; McCain, C.M.; Bowie, R.C.K.; Carnaval, A.C.; Moritz, C.; Rahbek, C.; Roberts, T.E.; et al. Latitude, elevational climatic zonation and speciation in the New World vertebrates. Proc. R. Soc. B 2012, 279, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, G. Ecologically crucial features of climate in high tropical mountains. In High Altitude Tropical Biogeography; Vuilleumier, F., Monasterio, M., Eds.; Oxford University Press: Oxford, UK, 1986; pp. 11–45. [Google Scholar]
- Wootton, A.; Enríquez, P.L.; Navarrete-Gutiérrez, D. Regional patterns of vegetation, temperature, and rainfall trends in the coastal mountain range of Chiapas, Mexico. Atmósfera 2023, 36, 91–122. [Google Scholar] [CrossRef]
- Posso-Terranova, A.; Andrés, J. Complex niche divergence underlies lineage diversification in Oophaga poison frogs. J. Biogeogr. 2016, 43, 2002–2015. [Google Scholar] [CrossRef]
- Owen, J.G. Population and geographic variation of Peromyscus leucopus in relation to climatic factors. J. Mammal. 1989, 70, 98–109. [Google Scholar] [CrossRef]
- Pérez-Consuegra, S.G.; Vázquez-Domínguez, E. Intricate evolutionary histories in montane species: A phylogenetic window into craniodental discrimination in the Peromyscus mexicanus species group (Mammalia: Rodentia: Cricetidae). J. Zool. Syst. Evol. Res. 2017, 55, 57–72. [Google Scholar] [CrossRef]
- Medina, R.; Wogan, G.O.U.; Bi, K.; Termignoni-García, F.; Bernal, M.H.; Jaramillo-Correa, J.P.; Wang, I.J.; Vázquez-Domínguez, E. Phenotypic and genomic diversification with isolation by environment along elevational gradients in a neotropical treefrog. Mol. Ecol. 2021, 30, 4062–4076. [Google Scholar] [CrossRef]
- Patton, J.L.; Smith, M. mtDNA phylogeny of Andean mice: A test of diversification across ecological gradients. Evolution 1992, 46, 174–183. [Google Scholar] [CrossRef]
- Pérez-Consuegra, S.G.; Vázquez-Domínguez, E. Mitochondrial diversification of the Peromyscus mexicanus species group in Nuclear Central America: Biogeographic and taxonomic implications. J. Zool. Syst. Evol. Res. 2015, 53, 300–311. [Google Scholar] [CrossRef]
- Gutiérrez-García, T.A.; Vázquez-Domínguez, E. Biogeographically dynamic genetic structure bridging two continents in the monotypic Central American rodent Ototylomys phyllotis. Biol. J. Linn. Soc. 2012, 107, 593–610. [Google Scholar] [CrossRef]
- Ornelas, J.F.; Sosa, V.; Soltis, D.E.; Daza, J.M.; González, C.; Soltis, P.S.; Gutiérrez-Rodríguez, C.; Espinosa de los Monteros, A.; Castoe, T.A.; Bell, C.; et al. Comparative phylogeographic analyses illustrate the complex evolutionary history of threatened cloud forests of northern Mesoamerica. PLoS ONE 2013, 8, e56283. [Google Scholar] [CrossRef] [PubMed]
- Bagley, J.C.; Johnson, J.B. Phylogeography and biogeography of the lower Central American Neotropics: Diversification between two continents and between two seas. Biol. Rev. 2014, 89, 767–790. [Google Scholar] [CrossRef] [PubMed]
- Iturralde-Vinent, M.A. El origen paleogeográfico de la biota de Guatemala. In Biodiversidad de Guatemala; Cano, E.B., Ed.; Universidad del Valle de Guatemala: Ciudad de Guatemala, Guatemala, 2006; pp. 1–6. [Google Scholar]
- Ornelas, J.F.; Ruiz-Sánchez, E.; Sosa, V. Phylogeography of Podocarpus matudae (Podocarpaceae): Pre-Quaternary relicts in northern Mesoamerican cloud forests. J. Biogeogr. 2010, 37, 2384–2396. [Google Scholar] [CrossRef]
- Ramírez-Barahona, S.; Eguiarte, L.E. The role of glacial cycles in promoting genetic diversity in the Neotropics: The case of cloud forests during the Last Glacial Maximum. Ecol. Evol. 2013, 3, 725–738. [Google Scholar] [CrossRef]
- Mayle, F.E.; Burn, M.J.; Power, M.; Urrego, D.H. Vegetation and fire at the Last Glacial Maximun in tropical South America. In Past Climate Variability in South America and Surrounding Regions. From the Last Glacial Maximum to the Holocene; Vimeux, F., Silvestre, F., Khodri., M., Eds.; Springer: Paris, France, 2009; pp. 89–112. [Google Scholar] [CrossRef]
- Botero, C.A.; Weising, F.J.; Wright, J.; Rubenstein, D.R. Evolutionary tipping points in the capacity to adapt to environmental change. Proc. Natl. Acad. Sci. USA 2015, 112, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Leadly, P.; Gonzalez, A.; Obura, D.; Krug, C.B.; Londoño-Murcia, M.C.; Milette, K.L.; Radulovici, A.; Rankovic, A.; Shannon, L.J.; Archer, E.; et al. Achieving global biodiversity goals by 2050 requires urgent and integrated actions. One Earth 2022, 5, 597–603. [Google Scholar] [CrossRef]
- Musser, G.G.; Carleton, M.D. Superfamily Muroidea. In Mammal Species of the World: A Taxonomic and Geographic Reference; Wilson, D.E., Reeder, D., Eds.; The John Hopkins University Press: Baltimore, MD, USA, 2005; pp. 894–1531. [Google Scholar]
- Dawson, W.D. Peromyscine biogeography, Mexican topography and Pleistocene climatology. In Contribuciones Mastozoológicas en Homenaje a Bernardo Villa; Sánchez-Cordero, V., Medellín, R.A., Eds.; CONABIO/Instituto de Biología/Instituto de Ecología, UNAM: Ciudad de México, Mexico, 2005; pp. 145–156. [Google Scholar]
- Lorenzo, C.; Ticul-Álvarez, S.; Pérez, S.G.; Patton, J.M. Revision of the Chiapan deer mouse, Peromyscus zarhynchus, with the description of a new species. J. Mammal. 2016, 97, 910–918. [Google Scholar] [CrossRef]
- Bradley, R.D.; Núñez-Tabares, M.; Soniat, T.J.; Kerr, S.; Raymond, R.W.; Ordóñez-Garza, N. Molecular Systematics and Phylogeography of Peromyscus nudipes (Cricetidae: Neotominae); Special Publications, Museum of Texas Tech University: Lubbock, TX, USA, 2016; Volume 65, pp. 201–213. [Google Scholar]
- Álvarez-Castañeda, S.T.; Lorenzo, C.; Segura-Trujillo, C.A.; Pérez-Consuegra, S.G. Two new species of Peromyscus from Chiapas, Mexico, and Guatemala. In From Field to Laboratory: A Memorial Volume in Honor of Robert J. Baker; Bradley, R.D., Genoways, H.H., Schmidly, D.J., Bradley, L.C., Eds.; Special Publications, Museum of Texas Tech University: Lubbock, TX, USA, 2019; pp. 543–558. [Google Scholar]
- Hernández-Canchola, G.; León-Paniagua, L.; Esselstyn, J.A. Paraphyletic relationships revealed by mitochondrial DNA in the Peromyscus mexicanus species group (Rodentia: Cricetidae). Rev. Mex. Biodiv. 2022, 93, e933811. [Google Scholar] [CrossRef]
- Flores-Villela, O.; Goyenechea, I. A comparison of hypotheses of historical area relationships for Mexico and Central America, or in search for the lost pattern. In Mesoamerican Herpetology: Systematics, Zoogeography, and Conservation; Johnson, J., Webb, R., Flores-Villela, O., Eds.; University of Texas: Austin, TX, USA, 2001; pp. 171–181. [Google Scholar]
- Jansa, S.A.; Giarla, T.C.; Lim, B.K. The phylogenetic position of the rodent genus Typhlomys and the geographic origin of Muroidea. J. Mammal. 2009, 90, 1083–1094. [Google Scholar] [CrossRef]
- Stephens, M.; Donnelly, P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2007, 73, 1162–1169. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Ersts, P.J. Geographic Distance Matrix Generator (v.1.2.3). American Museum of Natural History, Center for Biodiversity and Conservation. 2015. Available online: http://biodiversityinformatics.amnh.org/open_source/gdmg (accessed on 20 March 2023).
- Jensen, J.L.; Bohonak, A.J.; Kelley, S.T. Isolation by distance, web service. BMC Genet. 2005, 6, 13. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Engel, S.R.; Hogan, K.M.; Taylor, J.F.; Davis, S.K. Molecular systematics and paleobiogeography of the South American Sigmodontine Rodents. Mol. Biol. Evol. 1998, 15, 35–49. [Google Scholar] [CrossRef]
- León-Paniagua, L.; Navarro-Sigüenza, A.G.; Hernández-Baños, B.E.; Morales, J.C. Diversification of the arboreal mice of the genus Habromys (Rodentia: Cricetidae: Neotominae) in the Mesoamerican highlands. Mol. Phylogenet. Evol. 2007, 42, 653–664. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Li, W. Statistical test of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y. Statistical test of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Sánchez-Del Barrio, J.C.; Messenguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2597. [Google Scholar] [CrossRef]
- Fick, S.E.; Hijmans, R.J. Worldclim 2: New 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 2017, 37, 4302–4315. [Google Scholar] [CrossRef]
- IPCC. Emissions Scenarios; Nakicenovic, N., Swart, R., Eds.; Cambridge University Press: Cambridge, UK, 2000; Available online: https://www.ipcc.ch/report/emissions-scenarios (accessed on 20 March 2023).
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- Anderson, R.P.; González, I., Jr. Species-specific tuning increases robustness to sampling bias in models of species distributions: An implementation with Maxent. Ecol. Model. 2011, 222, 2796–2811. [Google Scholar] [CrossRef]
- Pearson, R.G.; Raxworthy, C.J.; Nakamura, M.; Peterson, A.T. Predicting species distributions from small numbers of occurrence records: A test case using cryptic geckos in Madagascar. J. Biogeogr. 2007, 34, 102–117. [Google Scholar] [CrossRef]
- Barraclough, T.G. How do species interactions affect evolutionary dynamics across whole communities? An. Rev. Ecol. Evol. Syst. 2015, 46, 25–48. [Google Scholar] [CrossRef]
- Kalkvik, H.M.; Stout, I.J.; Doonan, T.J.; Parkinson, C.L. Investigating niche and lineage diversification in widely distributed taxa: Phylogeography and ecological niche modeling of the Peromyscus maniculatus species group. Ecography 2012, 35, 54–64. [Google Scholar] [CrossRef]
- Urban, M.C.; Strauss, S.Y.; Pelletier, F.; Palkovacs, E.P.; Leibold, M.A.; Hendry, A.P.; De Meester, L.; Carlson, S.M.; Angert, A.L.; Giery, S.T. Evolutionary origins for ecological patterns in space. Proc. Natl. Acad. Sci. USA 2020, 117, 17482–17490. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.D.; Durish, N.D.; Rogers, D.S.; Miller, J.R.; Engstrom, M.D.; Kilpatrick, C.W. Toward a molecular phylogeny for Peromyscus: Evidence from mitochondrial cytochrome b sequences. J. Mammal. 2007, 88, 1146–1159. [Google Scholar] [CrossRef] [PubMed]
- Cronin, T.M. Paleoclimates, Understanding Climate Change Past and Present; Columbia University Press: New York, NY, USA, 2010. [Google Scholar]
- Daza, J.M.; Castoe, T.A.; Parkinson, C.L. Using regional comparative phylogeographic data from snake lineages to infer historical processes in Middle America. Ecography 2010, 33, 343–354. [Google Scholar] [CrossRef]
- Gutiérrez-García, T.A.; Vázquez-Domínguez, E. Consensus between genes and stones in the biogeographic and evolutionary history of Central America. Quat. Res. 2013, 79, 311–324. [Google Scholar] [CrossRef]
- Mendoza, A.; Bolívar-García, W.; Vázquez-Domínguez, E.; Ibañez, R.; Parra-Olea, G. The role of Central American barriers in shaping the evolutionary history of the northernmost glassfrog, Hyalinobatrachium fleischmanni (Anura: Centrolenidae). PeerJ 2019, 7, e6115. [Google Scholar] [CrossRef]
- Bush, M.B.; Corea-Metrio, A.Y.; Hodell, D.A.; Brenner, M.; Anselmetti, F.S.; Ariztegui, D.; Mueller, A.D.; Curtis, J.H.; Grzesik, D.A.; Burton, C.; et al. Re-evaluation of climate change in lowland Central America during the Last Glacial Maximum using new sediment cores from Lake Petén Itzá, Guatemala. In Past Climate Variability in South America and Surrounding Regions. From the Last Glacial Maximum to the Holocene; Vimeux, F., Silvestre, F., Khodri, M., Eds.; Springer: Paris, France, 2009; pp. 113–128. [Google Scholar] [CrossRef]
- McDonald, H.G.; Dávila, S.L. Mammoths in Central America: New records from Guatemala. Quat. Int. 2017, 443, 122–128. [Google Scholar] [CrossRef]
- Hillesheim, M.B.; Hodell, D.A.; Leyden, B.W.; Brenner, M.; Curtis, J.H.; Anselmentti, F.S.; Ariztegui, D.; Buck, D.G.; Guilderson, T.P.; Rosenmeier, M.F.; et al. Climate change in lowland Central America during the late deglacial and early Holocene. J. Quat. Sci. 2005, 20, 363–376. [Google Scholar] [CrossRef]
- Kappelle, M. Los Bosques de Roble (Quercus) de la Cordillera de Talamanca, Costa Rica: Biodiversidad, Ecología, Conservación y Desarrollo; Instituto Nacional de Biodiversidad/Universidad de Amsterdam: Heredia, Costa Rica, 1996. [Google Scholar]
- Yousefi, M.; Ahmadi, M.; Nourani, E.; Behrooz, R.; Rajabizadeh, M.; Geniez, P.; Kaboli, M. Upward altitudinal shifts in habitat suitability of mountain vipers since the Last Glacial Maximum. PLoS ONE 2015, 10, e0138087. [Google Scholar] [CrossRef]
- Qu, Y.; Luo, X.; Zhang, R.; Song, G.; Zou, F.; Lei, F. Lineage diversification and historical demography of a montane bird Garrulax elliotii—Implications for the Pleistocene evolutionary history of the eastern Himalayas. BMC Evol. Biol. 2011, 11, 174. [Google Scholar] [CrossRef]
- Mellick, R.; Wilson, P.D.; Rossetto, M. Demographic history and niche conservatism of tropical trees separated along an altitudinal gradient of a biogeographic barrier. Aust. J. Bot. 2014, 62, 438–450. [Google Scholar] [CrossRef]
- Shmueli, G. To explain or to predict? Stat. Sci. 2010, 25, 289–310. [Google Scholar] [CrossRef]
- Estrada, F.; Calderón-Bustamante, O.; Botzen, W.; Velasco, J.A.; Tol, R.S.J. AIRCC-Clim: A user-friendly tool for generating regional probabilistic climate change scenarios and risk measures. Environ. Model. Softw. 2022, 157, 105528. [Google Scholar] [CrossRef]
- Parmesan, C.; Yohe, G. A globally coherent fingerprint of climate change impacts across natural systems. Nature 2003, 421, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Plumptre, A.J.; Baisero, D.; Belote, R.T.; Vázquez-Domínguez, E.; Faurby, S.; Jêdrzejewski, W.; Kiara, H.; Kühl, H.; Benítez-López, A.; Luna-Aranguré, C.; et al. Where might we find ecologically intact communities? Front. For. Glob. Chang. 2021, 4, 626635. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage | Scenario A2 | Scenario B1 | |||||
|---|---|---|---|---|---|---|---|
| Current | 2020 | 2050 | 2080 | 2020 | 2050 | 2080 | |
| P. mexicanus | 8246 | 5620 | 3939 | 5738 | 2298 | 2956 | 2656 |
| +68% | +48% | +70% | −28% | +36% | +32% | ||
| P. gymnotis | 51,063 | 27,728 | 24,478 | 16,750 | 22,391 | 35,263 | 1606 |
| −54% | −48% | −33% | −44% | −69% | +3% | ||
| P. nicaraguae | 59,521 | 48,216 | 40,455 | 35,165 | 29,586 | 31,921 | 39,207 |
| −81% | −68% | −59% | −50% | −54% | −66% | ||
| P. guatemalensis | 12,918 | 3759 | 2861 | 3439 | 2598 | 5220 | 261 |
| −29% | −22% | −27% | −20% | −40% | −2% | ||
| P. salvadorensis | 17,613 | 14,424 | 24,285 | 1705 | 8.965 | 10,995 | 9178 |
| +82% | +138% | +10% | +51% | +62% | +52% | ||
| P. zarhynchus | 3095 | 622 | 184 | 775 | 174 | 809 | 620 |
| −20% | −6% | +25% | +6% | −26% | +20% | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Consuegra, S.G.; Sánchez-Tovar, L.; Rodríguez-Tapia, G.; Castañeda-Rico, S.; Vázquez-Domínguez, E. Late Pleistocene Altitudinal Segregation and Demography Define Future Climate Change Distribution of the Peromyscus mexicanus Species Group: Conservation Implications. Animals 2023, 13, 1753. https://doi.org/10.3390/ani13111753
Pérez-Consuegra SG, Sánchez-Tovar L, Rodríguez-Tapia G, Castañeda-Rico S, Vázquez-Domínguez E. Late Pleistocene Altitudinal Segregation and Demography Define Future Climate Change Distribution of the Peromyscus mexicanus Species Group: Conservation Implications. Animals. 2023; 13(11):1753. https://doi.org/10.3390/ani13111753
Chicago/Turabian StylePérez-Consuegra, Sergio G., Laura Sánchez-Tovar, Gerardo Rodríguez-Tapia, Susette Castañeda-Rico, and Ella Vázquez-Domínguez. 2023. "Late Pleistocene Altitudinal Segregation and Demography Define Future Climate Change Distribution of the Peromyscus mexicanus Species Group: Conservation Implications" Animals 13, no. 11: 1753. https://doi.org/10.3390/ani13111753