

First Description of the Mitogenome Features of Neofoleyellides Genus (Nematoda: Onchocercidae) Isolated from a Wild Bird (Pyrrhocorax pyrrhocorax)
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, Total DNA Extraction and Molecular Detection
2.2. Construction of the Genomic Library and Sequencing
2.3. Genomic Assembly
2.4. Annotation and Bioinformatic Analysis
2.5. Phylogenetic Analysis
3. Results and Discussion
3.1. Acquisition of 18S rDNA and Mitochondrial cox1 Genes
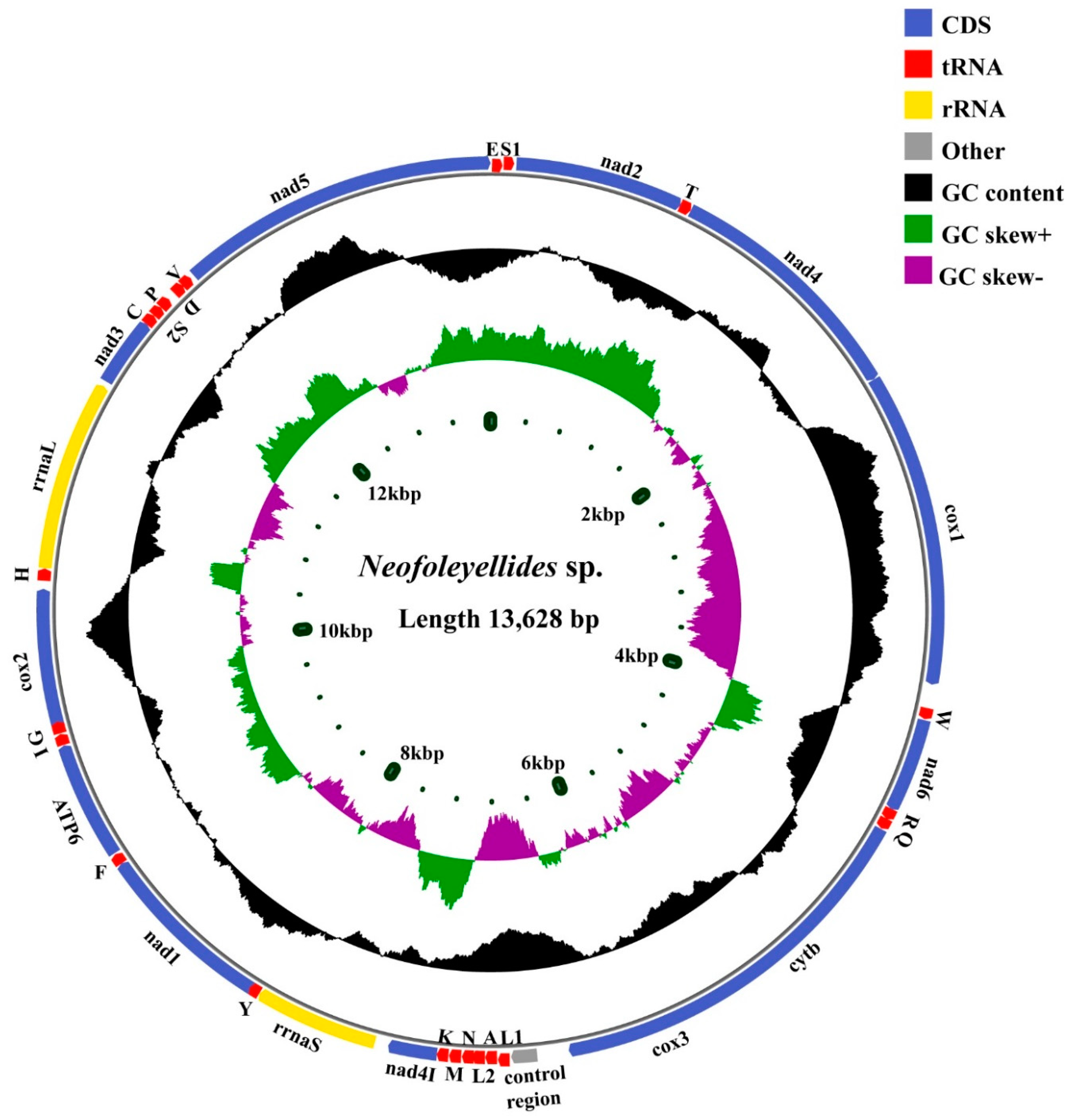
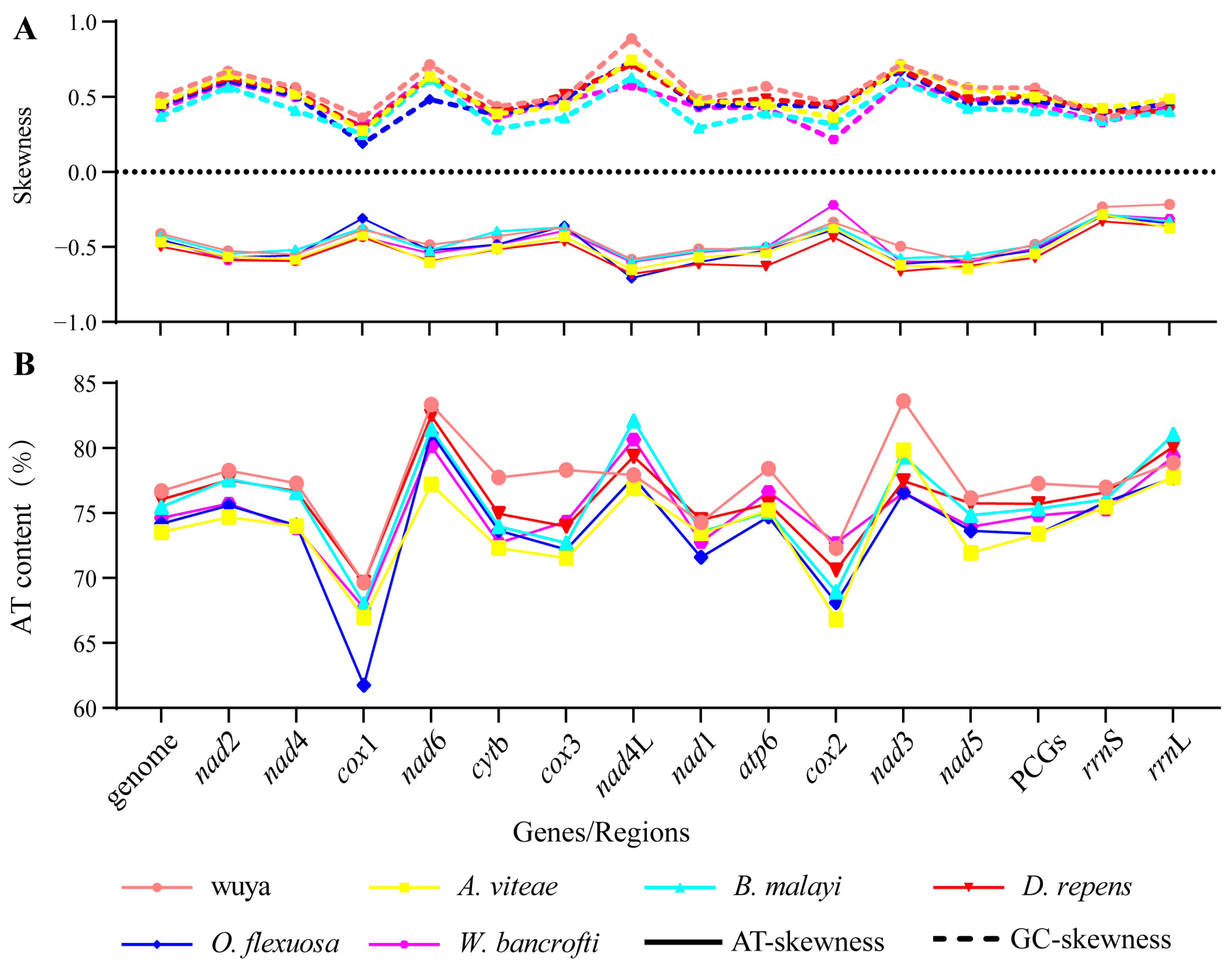
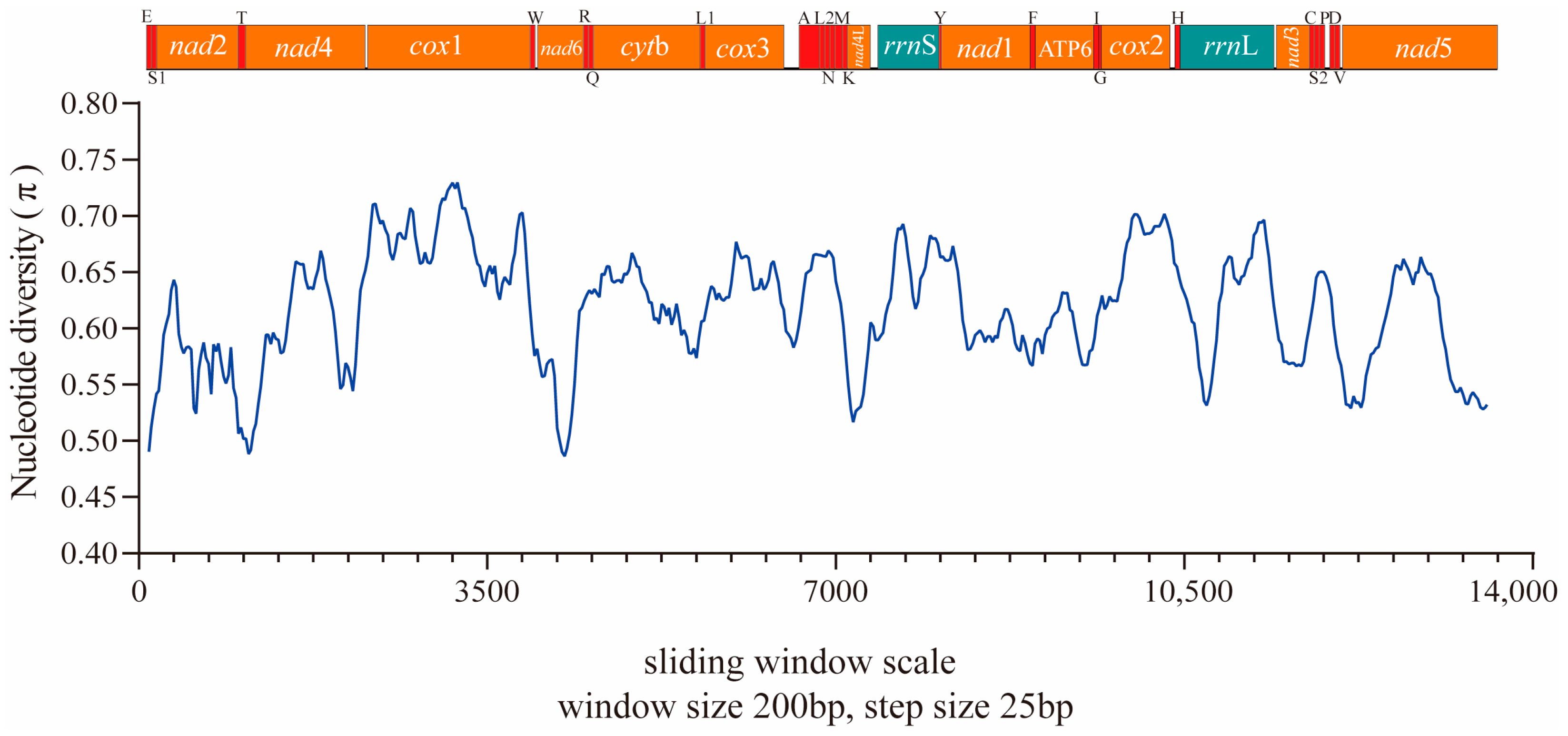
3.2. Mitogenome Organization and Composition
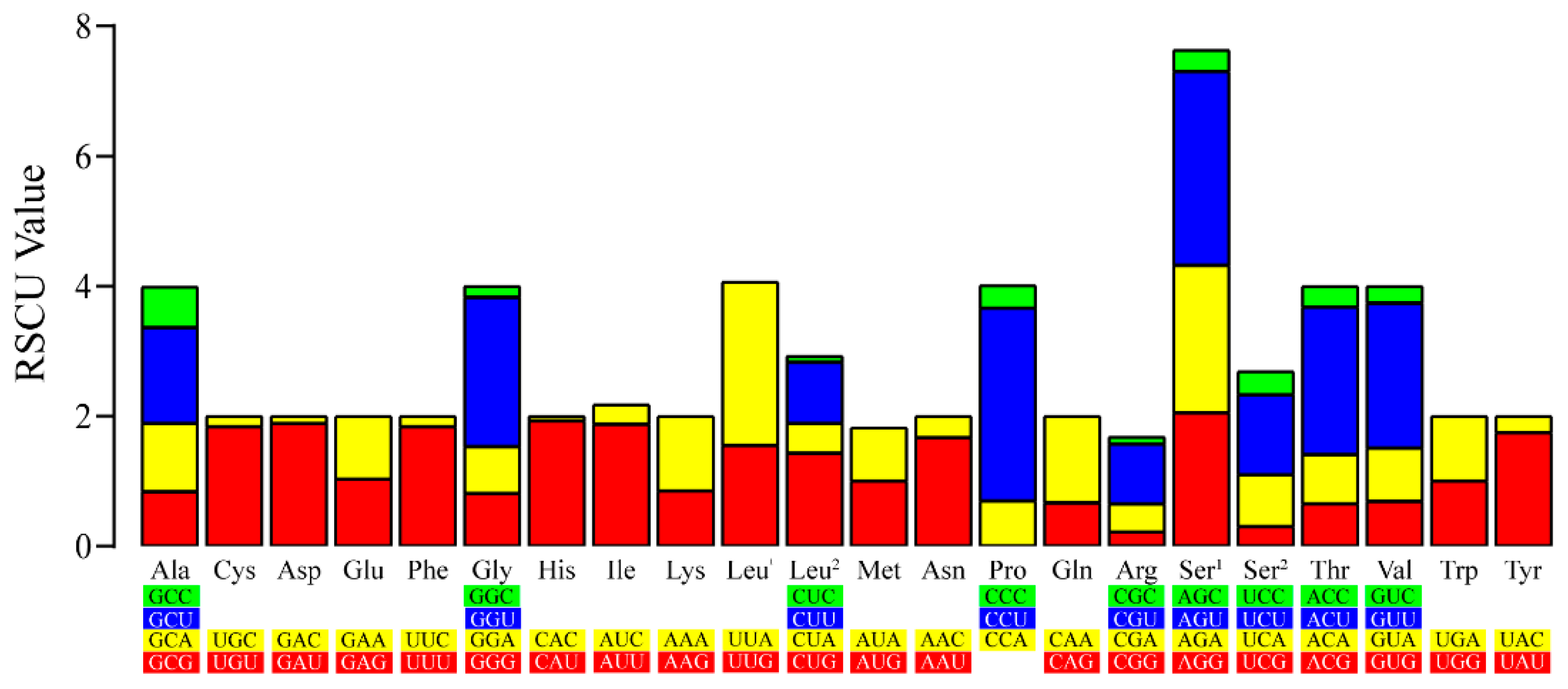
3.3. Characteristics of PCGs
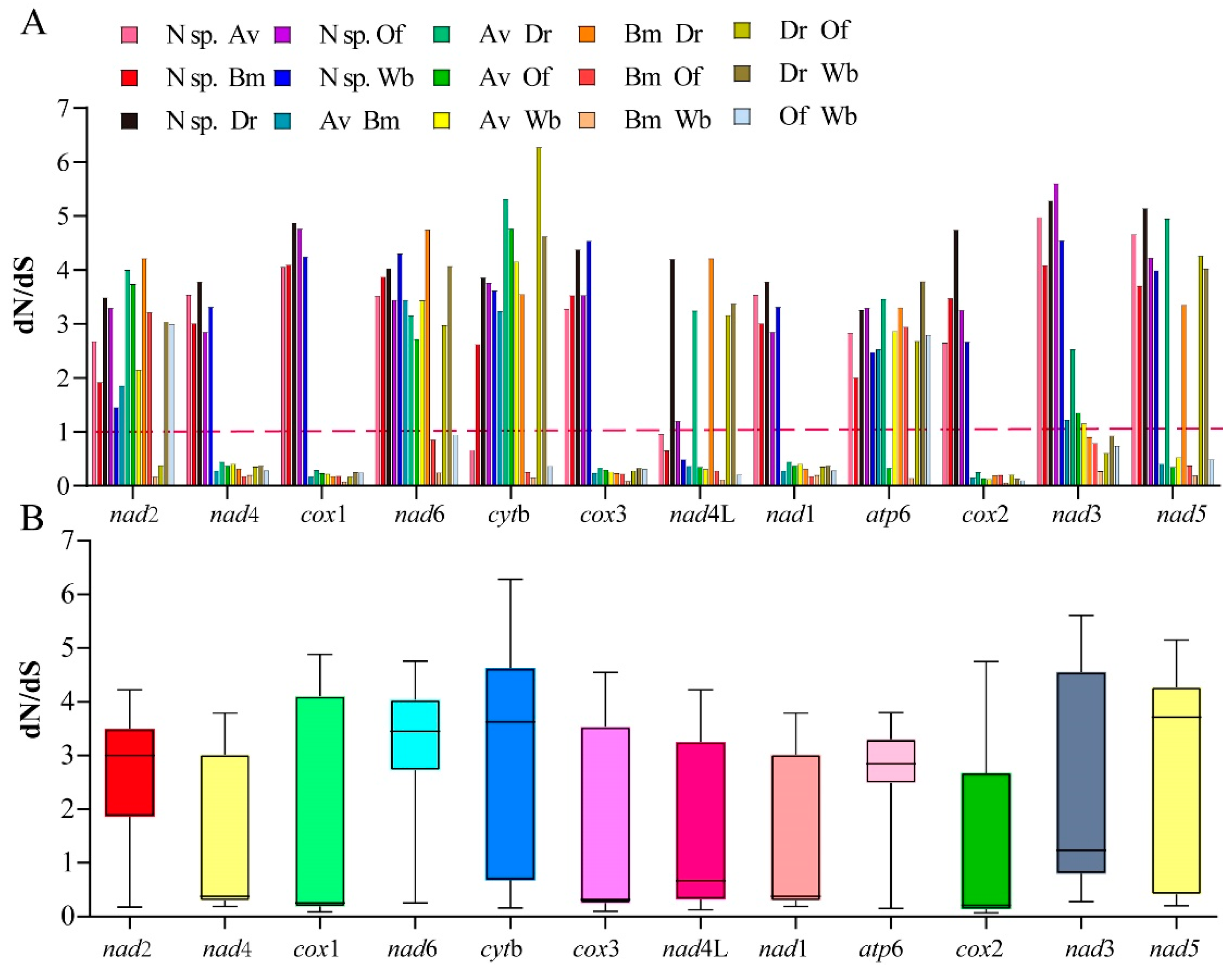
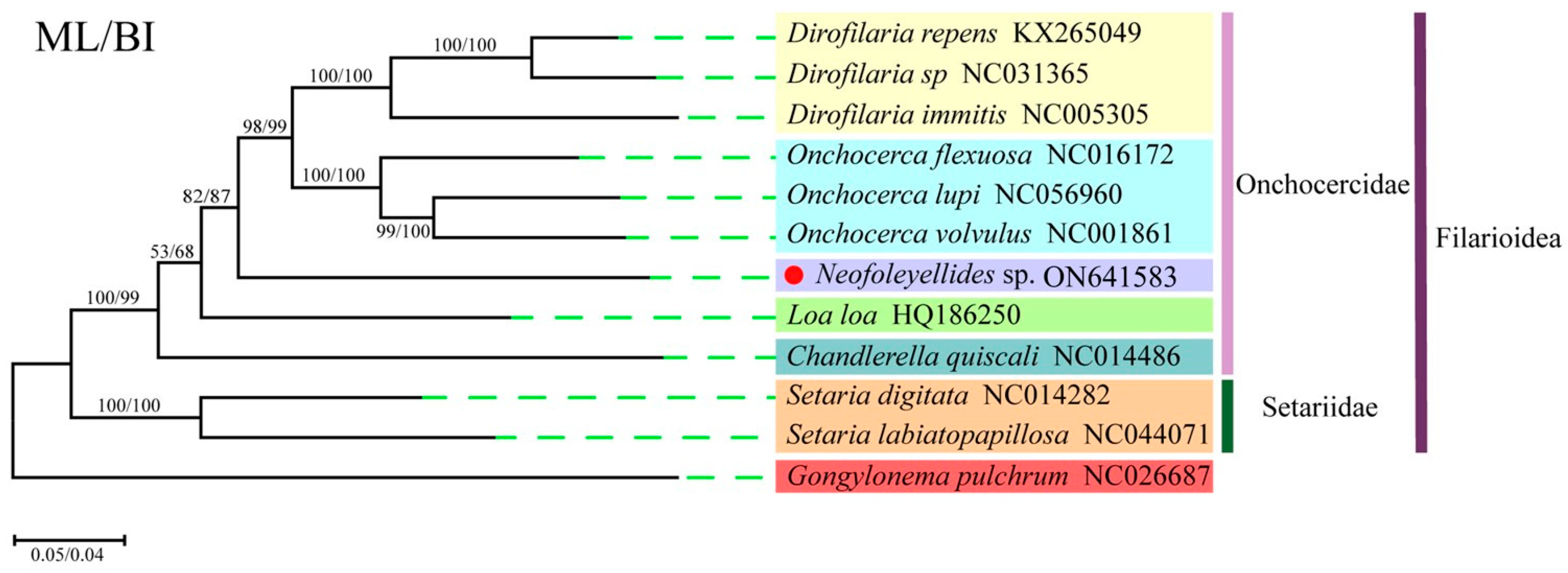
3.4. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martin, C.; Bain, O.; Jouvenet, N.; Raharimanga, V.; Robert, V.; Rousset, D. First report of Litomosa spp. (Nematoda: Filarioidea) from Malagasy bats; review of the genus and relationships between species. Parasite 2006, 13, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bain, O.; Otranto, D.; Diniz, D.G.; dos Santos, J.N.; de Oliveira, N.P.; Frota de Almeida, I.N.; de Almeida, R.N.F.; de Almeida, L.N.F.; Dantas-Torres, F.; de Almeida Sobrinho, E.F. Human intraocular filariasis caused by Pelecitus sp. nematode, Brazil. Emerg. Infect. Dis. 2011, 17, 867–869. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.F.; Earlé, R.A.; Du Toit, H.; Huchzermeyer, F.W. A host-parasite catalogue of the haematozoa of the sub-Saharan birds. Onderstepoort. J. Vet. Res. 1992, 59, 1–73. [Google Scholar] [PubMed]
- Mat Udin, A.S.; Uni, S.; Zainuri, N.A.; Abdullah Halim, M.R.; Belabut, D.A. Morphological characteristics of microfilariae in blood smears of the common treeshrew Tupaia glis (Mammalia: Scandentia) in Gemas, Negeri Sembilan, Malaysia. Trop. Biomed. 2020, 37, 1152–1157. [Google Scholar]
- Lefoulon, E.; Bain, O.; Bourret, J.; Junker, K.; Guerrero, R.; Cañizales, I.; Kuzmin, Y.; Satoto, T.B.T.; Cardenas-Callirgos, J.M.; Lima, S.D.S.; et al. Shaking the tree: Multi-locus sequence typing usurps current onchocercid (filarial nematode) phylogeny. PLoS Negl. Trop. Dis. 2015, 9, e0004233. [Google Scholar] [CrossRef]
- Roe, C.C.; Yaglom, H.; Howard, A.; Urbanz, J.; Verocai, G.G.; Andrews, L.; Harrison, V.; Barnes, R.; Lyons, T.; Bowers, J.R.; et al. Coyotes as reservoirs for Onchocerca lupi, United States, 2015–2018. Emerg. Infect. Dis. 2020, 26, 2989–2993. [Google Scholar] [CrossRef]
- Bain, O.; Casiraghi, M.; Martin, C.; Uni, S. The Nematoda Filarioidea: Critical analysis linking molecular and traditional approaches. Parasite 2008, 15, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Kuzmin, Y.; Netherlands, E.C.; du Preez, L.H.; Svitin, R. Two new species of Neofoleyellides (Nematoda: Onchocercidae) parasitising anuran amphibians in South Africa. Int. J. Parasitol. Parasites. Wildl. 2021, 14, 298–307. [Google Scholar] [CrossRef]
- Muñoz-García, C.I.; López-Díaz, O.; Osorio-Sarabia, D.; Martínez-Hernández, F.; Villalobos, G.; Isaak-Delgado, A.B.; Rendón-Franco, E.; Carreño-Cervantes, A.; Contreras-Patiño, D.R.; Berriatua, E.; et al. New insights into the clinico-histopathological and molecular features of Pelecitus (Filarioidea: Onchocercidae) from a raptor bird. Parasitol. Res. 2018, 117, 3319–3325. [Google Scholar] [CrossRef]
- Murugan, K.; Vadivalagan, C.; Karthika, P.; Panneerselvam, C.; Paulpandi, M.; Subramaniam, J.; Wei, H.; Aziz, A.T.; AlSalhi, M.; Devanesan, S.; et al. DNA barcoding and molecular evolution of mosquito vectors of medical and veterinary importance. Parasitol. Res. 2016, 115, 107–121. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Y.; Yang, X.; Qiu, J.-H.; Duan, H.; Xu, W.-W.; Chang, Q.-C.; Wang, C.-R. Mitochondrial DNA evidence supports the hypothesis that Triodontophorus species belong to Cyathostominae. Front. Microbiol. 2017, 8, 1444. [Google Scholar] [CrossRef] [PubMed]
- Shragai, T.; Tesla, B.; Murdock, C.; Harrington, L.C. Zika and chikungunya: Mosquito-borne viruses in a changing world. Ann. N. Y. Acad. Sci. 2017, 1399, 61–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binkienė, R.; Chagas, C.R.F.; Bernotienė, R.; Valkiūnas, G. Molecular and morphological characterization of three new species of avian Onchocercidae (Nematoda) with emphasis on circulating microfilariae. Parasit. Vectors. 2021, 14, 137. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome. Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. J. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Burland, T.G. DNASTAR’s Lasergene sequence analysis software. J. Methods Mol. Biol. 2000, 132, 71–91. [Google Scholar]
- Do Nascimento, B.L.S.; Da Silva, F.S.; Nunes-Neto, J.P.; De Almeida Medeiros, D.B.; Cruz, A.C.R.; Da Silva, S.P.; da Silva e Silva, L.H.; de Oliveira Monteiro, H.A.; Dias, D.D.; Vieira, D.B.R.; et al. First description of the mitogenome and phylogeny of Culicinae species from the Amazon region. Genes 2021, 12, 1983. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, S.; Kang, J.; Fang, H.; Dao, H.; Guo, W.; Lai, C.; Lai, M.; Fan, J.; Fu, L.; et al. Evolving molecular epidemiological profile of human immunodeficiency virus 1 in the southwest border of China. PLoS ONE 2014, 9, e10757824. [Google Scholar] [CrossRef] [Green Version]
- Netherlands, E.C.; Svitin, R.; Cook, C.A.; Smit, N.J.; Brendonck, L.; Vanhove, M.; Du Preez, L.H. Neofoleyellides boerewors n. gen. n. sp. (Nematoda: Onchocercidae) parasitising common toads and mosquito vectors: Morphology, life history, experimental transmission and host-vector interaction in situ. Int. J. Parasitol. 2020, 50, 177–194. [Google Scholar] [CrossRef]
- Da Silva, A.F.; Machado, L.C.; De Paula, M.B.; Vieira, C.J.D.S.P.; Bronzoni, R.V.D.M.; Santos, M.A.V.D.M.; Wallau, G.L. Culicidae evolutionary history focusing on the culicinae subfamily based on mitochondrial phylogenomics. Sci. Rep. 2020, 10, 18823. [Google Scholar] [CrossRef]
- Liu, G.-H.; Gasser, R.B.; Otranto, D.; Xu, M.-J.; Shen, J.-L.; Mohandas, N.; Zhou, D.-H.; Zhu, X.-Q. Mitochondrial genome of the eyeworm, Thelazia callipaeda (Nematoda: Spirurida), as the first representative from the family Thelaziidae. PLoS Negl. Trop. Dis. 2013, 7, e2029. [Google Scholar] [CrossRef] [Green Version]
- Hardy, C.M.; Court, L.N.; Morgan, M.J.; Webb, C.E. The complete mitochondrial DNA genomes for two lineages of Aedes notoscriptus (Diptera: Culicidae). Mitochondrial. DNA A DNA Mapp. Seq. Anal. 2016, 27, 2024–2025. [Google Scholar]
- Matthews, B.J.; Dudchenko, O.; Kingan, S.B.; Koren, S.; Antoshechkin, I.; Crawford, J.E.; Glassford, W.J.; Herre, M.; Redmond, S.N.; Rose, N.H.; et al. Improved reference genome of Aedes aegypti informs arbovirus vector control. Nature 2018, 563, 501–507. [Google Scholar] [CrossRef]
- Bang, W.J.; Won, M.H.; Cho, S.T.; Ryu, J.; Choi, K.S. A multiplex PCR assay for six Aedini species, including Aedes albopictus. Parasit. Vectors 2021, 14, 380. [Google Scholar] [CrossRef]
- Yatawara, L.; Wickramasinghe, S.; Rajapakse, R.P.; Agatsuma, T. The complete mitochondrial genome of Setaria digitata (Nematoda: Filarioidea): Mitochondrial gene content, arrangement and composition compared with other nematodes. Mol. Biochem. Parasitol. 2010, 173, 32–38. [Google Scholar] [CrossRef]
- Luo, Q.-C.; Hao, Y.-J.; Meng, F.; Li, T.-J.; Ding, Y.-R.; Hua, Y.-Q.; Bin Chen, B. The mitochondrial genomes of Culex tritaeniorhynchus and Culex pipiens pallens (Diptera: Culicidae) and comparison analysis with two other Culex species. Parasit. Vectors. 2016, 9, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, C.C.; Urbanz, J.; Andrews, L.; Verocai, G.G.; Engelthaler, D.M.; Hepp, C.M.; Sahl, J.W. Complete mitochondrial genome of Onchocerca lupi (Nematoda, Onchocercidae). Mitochondrial. DNA B Resour. 2021, 6, 2572–2574. [Google Scholar] [CrossRef] [PubMed]
- Beckenbach, A.T.; Stewart, J.B. Insect mitochondrial genomics 3: The complete mitochondrial genome sequences of representatives from two neuropteroid orders: A dobsonfly (order Megaloptera) and a giant lacewing and an owlfly (order Neuroptera). Genome 2009, 52, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Behura, S.K.; Severson, D.W. Codon usage bias: Causative factors, quantification methods and genome-wide patterns: With emphasis on insect genomes. Biol. Rev. Camb. Philos. Soc. 2013, 88, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.-J.; Zou, Y.-L.; Ding, Y.-R.; Xu, W.-Y.; Yan, Z.-T.; Li, X.-D.; Fu, W.-B.; Li, T.-J.; Chen, B. Complete mitochondrial genomes of Anopheles stephensi and An. dirus and comparative evolutionary mitochondriomics of 50 mosquitoes. Sci. Rep. 2017, 7, 7666. [Google Scholar] [CrossRef]
- Meganathan, P.R.; Dubey, B.; Batzer, M.A.; Ray, D.A.; Haque, I. Complete mitochondrial genome sequences of three Crocodylus species and their comparison within the Order Crocodylia. Gene 2011, 478, 35–41. [Google Scholar] [CrossRef]
- Li, H.; Liu, H.; Shi, A.; Stys, P.; Zhou, X.; Cai, W. The complete mitochondrial genome and novel gene arrangement of the unique-headed bug Stenopirates sp. (Hemiptera: Enicocephalidae). PLoS ONE 2012, 7, e29419. [Google Scholar] [CrossRef] [Green Version]
- Traversa, D.; Costanzo, F.; Iorio, R.; Aroch, I.; Lavy, E. Mitochondrial cytochrome c oxidase subunit 1 (cox1) gene sequence of Spirocerca lupi (Nematoda, Spirurida): Avenues for potential implications. Vet. Parasitol. 2007, 146, 263–270. [Google Scholar] [CrossRef]
- Weeraratne, T.C.; Surendran, S.N.; Parakrama Karunaratne, S.H.P. DNA barcoding of morphologically characterized mosquitoes belonging to the subfamily Culicinae from Sri Lanka. Parasit. Vectors. 2018, 11, 266. [Google Scholar] [CrossRef] [Green Version]
- Small, S.T.; Tisch, D.J.; Zimmerman, P.A. Molecular epidemiology, phylogeny and evolution of the filarial nematode Wuchereria bancrofti. Infect. Genet. Evol. 2014, 28, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Ferreira de Freitas, L.; Bartholomay, L.C. The Taxonomic History of Ochlerotatus Lynch Arribálzaga, 1891 (Diptera: Culicidae). Insects 2021, 12, 452. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Strand | Position and Length (bp) | Initiation Codon | Stop Codon | Anticodon |
|---|---|---|---|---|---|
| trnE | N | 1–57 (57) | TTC | ||
| trnS1 | N | 58–108 (51) | TCT | ||
| nad2 | N | 108–944 (838) | TTG | TA(A) | |
| trnT | N | 944–999 (56) | TGT | ||
| nad4 | N | 1001–2224 (1224) | ATA | TAA | |
| cox1 | N | 2226–3878 (1653) | ATG | TAG | |
| trnW | N | 3879–3933 (55) | TCA | ||
| nad6 | N | 3939–4400 (462) | ATT | TAA | |
| trnR | N | 4399–4452 (54) | ACG | ||
| trnQ | N | 4452–4505 (54) | TTG | ||
| cytb | N | 4509–5592 (1084) | ATG | T(AA) | |
| trnL1 | N | 5592–5647 (56) | TAG | ||
| cox3 | N | 5644–6426 (783) | ATA | TAA | |
| trnA | N | 6777–6833 (57) | TGC | ||
| trnL2 | N | 6836–6895 (60) | TAA | ||
| trnN | N | 6890–6946 (57) | GTT | ||
| trnM | N | 6951–7009 (59) | CAT | ||
| trnK | N | 7013–7068 (56) | CTT | ||
| nad4L | N | 7069–7308 (240) | ATT | TAA | |
| rrnS | N | 7310–7985 (676) | ATA | TAA | |
| trnY | N | 7986–8039 (54) | GTA | ||
| nad1 | N | 8042–8911 (870) | TTG | TAG | |
| trnF | N | 8911–8964 (54) | GAA | ||
| atp6 | N | 8971–9549 (579) | ATG | TAA | |
| trnI | N | 9553–9607 (55) | GAT | ||
| trnG | N | 9612–9669 (58) | TCC | ||
| cox2 | N | 9669–10,358 (690) | ATT | TAG | |
| trnH | N | 10,358–10,416 (59) | GTG | ||
| rrnL | N | 10,418–11,386 (969) | ATT | T(AA) | |
| nad3 | N | 11,380–11,721 (342) | ATT | TAA | |
| trnC | N | 11,720–11,774(55) | GCA | ||
| trnS2 | N | 11,776–11,826(51) | TGA | ||
| trnP | N | 11,828–11,882 (55) | AGG | ||
| trnD | N | 11,922–11,976 (55) | GTC | ||
| trnV | N | 11,976–12,029 (54) | TAC | ||
| nad5 | N | 12,029–13,621 (1593) | ATT | TAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, T.; Ma, X.; Wang, F.; Xie, L.; Lv, Q.; Zeng, M.; Xu, Y.; Qin, S.; Chang, Q. First Description of the Mitogenome Features of Neofoleyellides Genus (Nematoda: Onchocercidae) Isolated from a Wild Bird (Pyrrhocorax pyrrhocorax). Animals 2022, 12, 2854. https://doi.org/10.3390/ani12202854
Wu T, Ma X, Wang F, Xie L, Lv Q, Zeng M, Xu Y, Qin S, Chang Q. First Description of the Mitogenome Features of Neofoleyellides Genus (Nematoda: Onchocercidae) Isolated from a Wild Bird (Pyrrhocorax pyrrhocorax). Animals. 2022; 12(20):2854. https://doi.org/10.3390/ani12202854
Chicago/Turabian StyleWu, Tingting, Xiaoxiao Ma, Fengfeng Wang, Linhong Xie, Qingbo Lv, Minhao Zeng, Yu Xu, Siyuan Qin, and Qiaocheng Chang. 2022. "First Description of the Mitogenome Features of Neofoleyellides Genus (Nematoda: Onchocercidae) Isolated from a Wild Bird (Pyrrhocorax pyrrhocorax)" Animals 12, no. 20: 2854. https://doi.org/10.3390/ani12202854