
Concerted and Independent Evolution of Control Regions 1 and 2 of Water Monitor Lizards (Varanus salvator macromaculatus) and Different Phylogenetic Informative Markers



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Collection and DNA Extraction
2.2. CR1 Sequencing
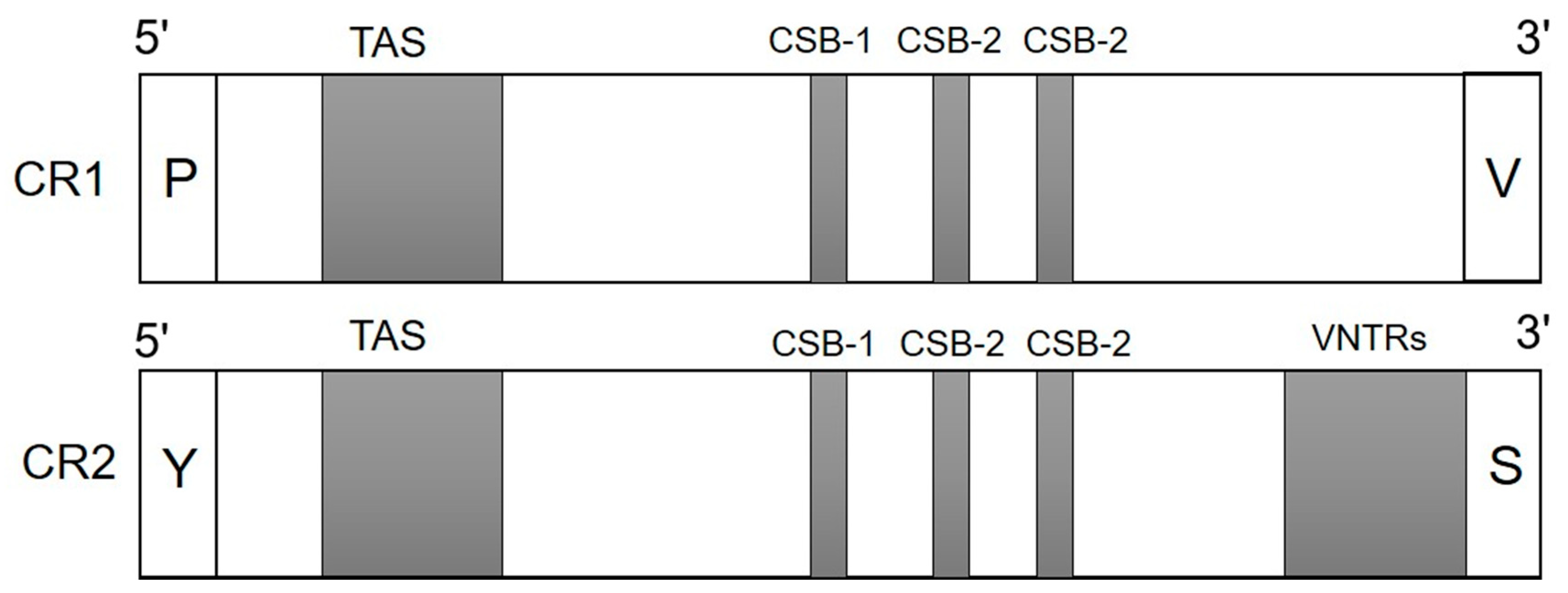
2.3. Positional Annotation
2.4. Comparison of Genetic Variability Based on CR Sequences at the Population Level
2.5. Phylogenetic Analysis Based on Mitochondrial CR1 and CR2 Sequences at the Species Level

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | GenBank Accession Number | CRs | Reference | |
|---|---|---|---|---|
| CR1 | CR2 | |||
| Varanus salvator macromaculatus | LC326253-LC326324 | CR1 | - | This study |
| Varanus salvator macromaculatus | LC326325-LC326396 | - | CR2 | Wongtienchai et al. [5] |
| Varanus salvator | EU747731 | CR1 | CR2 | Castoe et al. [36] |
| Varanus salvator macromaculatus | AB980995 | CR1 | CR2 | Chaiprasertsri et al. [37] |
| Varanus salvator komaini | AB980996 | CR1 | CR2 | Chaiprasertsri et al. [37] |
| Varanus exanthematicus | AB738957 | CR1 | CR2 | - |
| Varanus komodoensis | AB080276 | CR1 | CR2 | Kumazawa and Endo [11] |
| Varanus niloticus | AB185327 | CR1 | CR2 | Kumazawa [13] |
2.6. Recombination Testing
3. Results
3.1. Positional Annotation in the Control Regions of V. salvator macromaculatus
3.2. Sequence Variation in the CRs of V. salvator macromaculatus
3.3. Phylogenetic Relationship Based on the Control Regions of V. salvator macromaculatus
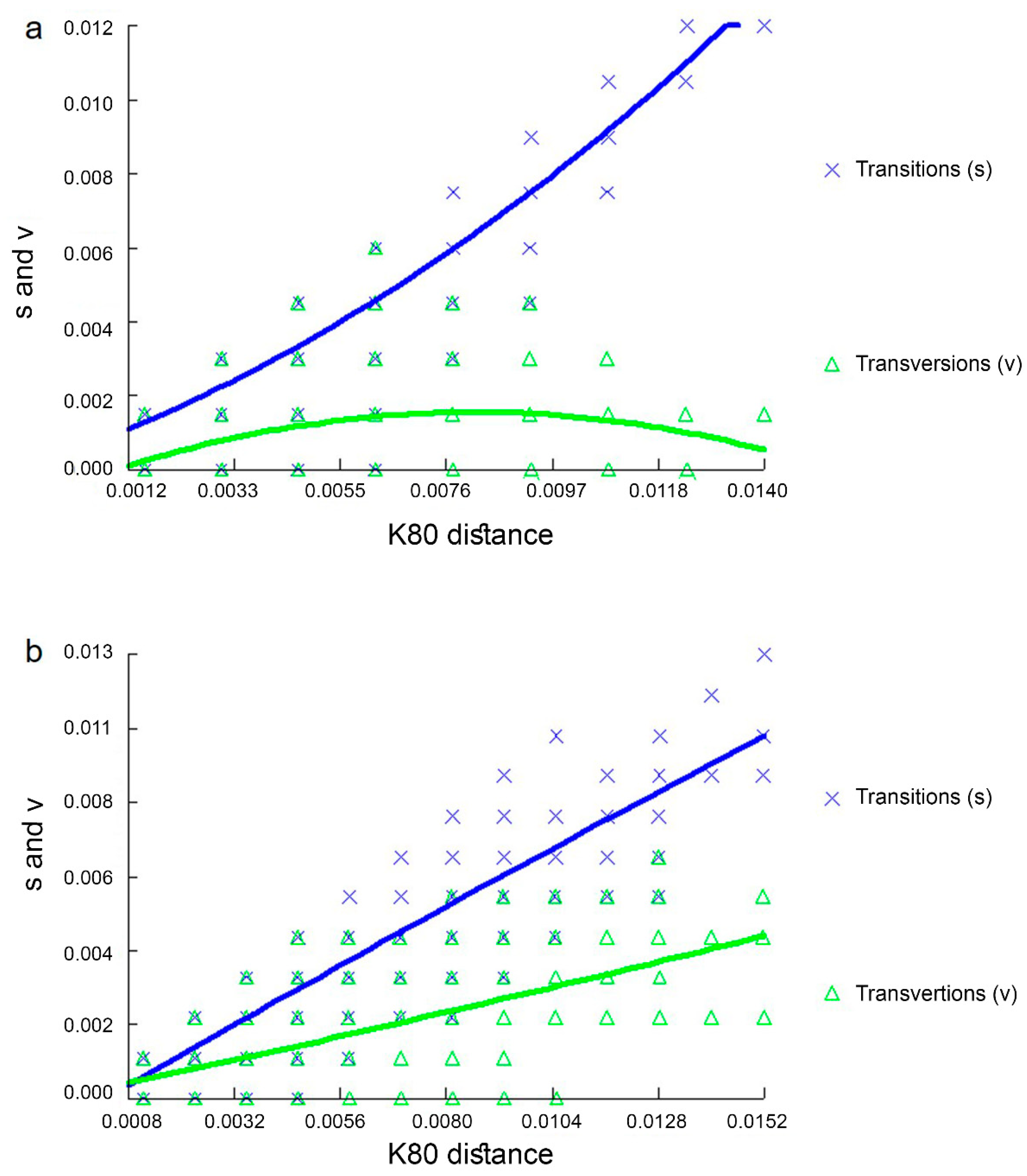
3.4. Recombination Events in CRs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, M.C.; Noguchi, E. Regulation of DNA replication through natural impediments in the eukaryotic genome. Genes 2017, 8, 98. [Google Scholar] [CrossRef] [Green Version]
- Areesirisuk, P.; Muangmai, N.; Kunya, K.; Singchat, W.; Sillapaprayoon, S.; Lapbenjakul, S.; Thapana, W.; Kantachumpoo, A.; Baicharoen, S.; Rerkamnuaychoke, B.; et al. Characterization of five complete Cyrtodactylus mitogenome structures reveals low structural diversity and conservation of repeated sequences in the lineage. PeerJ 2018, 6, e6121. [Google Scholar] [CrossRef] [PubMed]
- Vinson, C.C.; Mangaravite, E.; Sebbenn, A.M.; Lander, T.A. Using molecular markers to investigate genetic diversity, mating system and gene flow of Neotropical trees. Rev. Bras. Bot. 2018, 41, 481–496. [Google Scholar] [CrossRef]
- Wongtienchai, P.; Lapbenjakul, S.; Jangtarwan, K.; Areesirisuk, P.; Mahaprom, R.; Subpayakom, N.; Singchat, S.; Sillapaprayoon, S.; Muangmai, N.; Songchan, R.; et al. Genetic management of a water monitor lizard (Varanus salvator macromaculatus) population at Bang Kachao Peninsula as a consequence of urbanization with Varanus Farm Kamphaeng Saen as the first captive research establishment. J. Zool. Syst. Evol. Res. 2021, 59, 484–497. [Google Scholar] [CrossRef]
- Zheng, C.; Nie, L.; Wang, J.; Zhou, H.; Hou, H.; Wang, H.; Liu, J. Recombination and evolution of duplicate control regions in the mitochondrial genome of the Asian big-headed turtle, Platysternon megacephalum. PLoS ONE 2013, 8, e82854. [Google Scholar] [CrossRef] [PubMed]
- Singchat, W.; Areesirisuk, P.; Sillapaprayoon, S.; Muangmai, N.; Baicharoen, S.; Suntrarachun, S.; Chanhome, L.; Peyachoknagul, S.; Srikulnath, K. Complete mitochondrial genome of Siamese cobra (Naja kaouthia) determined using next-generation sequencing. Mitochondrial DNA Part B Resour. 2019, 4, 577–578. [Google Scholar] [CrossRef]
- Lallemand, T.; Leduc, M.; Landès, C.; Rizzon, C.; Lerat, E. An overview of duplicated gene detection methods: Why the duplication mechanism has to be accounted for in their choice. Genes 2020, 11, 1046. [Google Scholar] [CrossRef]
- Li, D.H.; Shi, W.; Munroe, T.A.; Gong, L.; Kong, X.Y. Concerted evolution of duplicate control regions in the mitochondria of species of the flatfish family Bothidae (Teleostei: Pleuronectiformes). PLoS ONE 2015, 10, e0134580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uetz, P.; Freed, P.; Aguilar, R.; Hošek, J. The Reptile Database. 2021. Available online: http://www.reptile-database.org (accessed on 23 May 2006).
- Ouwens, P.A. On a large Varanus species from the island of Komodo. Bull. Jard. Bot. Buitenzorg. 1912, 6, 1–3. [Google Scholar]
- Kumazawa, Y.; Endo, H. Mitochondrial genome of the Komodo dragon: Efficient sequencing method with reptile-oriented primers and novel gene rearrangements. DNA Res. 2004, 11, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Fitzinger, L. Neue Classification der Reptilien nach ihren natürlichen Verwandtschaften nebst einer Verwandschafts-Tafel und einem Verzeichnisse der Reptilien-Sammlung des K. K. Zoologischen Museums zu Wien; J.G., Heubner: Wien, Austria, 1826; p. 66. [Google Scholar]
- Kumazawa, Y. Mitochondrial genomes from major lizard families suggest their phylogenetic relationships and ancient radiations. Gene 2007, 388, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Amer, S.A.; Kumazawa, Y. Timing of a mtDNA gene rearrangement and intercontinental dispersal of varanid lizards. Genes Genet. Syst. 2008, 83, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Deraniyagala, P.E.P. Four New Races of the “Kabaragoya” Lizard Varanus salvator. Spolia Zeylan. 1944, 24, 59–62. [Google Scholar]
- Hague, M.T.J.; Routman, E.J. Does population size affect genetic diversity? A test with sympatric lizard species. J. Hered. 2016, 116, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Supikamolseni, A.; Ngaoburanawit, N.; Sumontha, M.; Chanhome, L.; Suntrarachun, S.; Peyachoknagul, S.; Srikulnath, K. Molecular barcoding of venomous snakes and species-specific multiplex PCR assay to identify snake groups for which antivenom is available in Thailand. Genet. Mol. Res. 2015, 14, 13981–13997. [Google Scholar] [CrossRef] [PubMed]
- Parham, J.F.; Feldman, C.R.; Boore, J.L. The complete mitochondrial genome of the enigmatic big-headed turtle (Platysternon): Description of unusual genomic features and the reconciliation of phylogenetic hypotheses based on mitochondrial and nuclear DNA. BMC Evol. Biol. 2006, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.L.; Nie, L.W.; Pu, Y.G. Complete mitochondrial genome of Chinese big-headed turtle, Platysternon megacephalum, with a novel gene organization in vertebrate mtDNA. Gene 2006, 380, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Easterling, K.A.; Pitra, N.J.; Morcol, T.B.; Aquino, J.R.; Lopes, L.G.; Bussey, K.C.; Matthews, P.D.; Bass, H.W. Identification of tandem repeat families from long-read sequences of Humulus lupulus. PLoS ONE 2020, 15, e0233971. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Clement, M.; Snell, Q.; Walker, P.; Posada, D.; Crandall, K. TCS: Estimating gene genealogies. 16th International Parallel and Distributed Processing Symposium. Int. Proc. 2002, 2, 184. [Google Scholar]
- Dalgaard, P. Power and the computation of sample size. In Introductory Statistics with R; Springer: New York, NY, USA, 2008; pp. 155–162. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Salemi, M. Nucleotide substitution models. PRACTICE: The PHYLIP and TREEPUZZLE software packages. In The Phylogenetic Handbook a Practical Approach to Phylogenetic Analysis and Hypothesis Testing; Salemi, M., Vandamme, A.M., Eds.; Cambridge University Press: Cambridge, UK, 2003; pp. 88–97. [Google Scholar]
- Felsenstein, J. Distance methods for inferring phylogenies: A justification. Evolution 1984, 38, 16–24. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Xia, X.; Lemey, P. Assessing Substitution Saturation with DAMBE. The Phylogenetic Handbook: A Practical Approach to DNA and Protein Phylogeny; Cambridge University Press: Cambridge, UK, 2009; pp. 615–630. [Google Scholar]
- Xia, X. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Sys. Biol. 2018, 67, 901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanabe, A.S. Kakusan4 and Aminosan: Two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Mol. Ecol. Resour. 2011, 11, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Castoe, T.A.; Jiang, Z.J.; Gu, W.; Wang, Z.O.; Pollock, D.D. Adaptive evolution and functional redesign of core metabolic proteins in snakes. PLoS ONE 2008, 3, e2201. [Google Scholar] [CrossRef] [Green Version]
- Chaiprasertsri, N.; Uno, Y.; Peyachoknagul, S.; Prakhongcheep, O.; Baicharoen, S.; Charernsuk, S.; Nishida, C.; Mutsuda, Y.; Koga, A.; Srikulnath, K. Highly species-specific centromeric repetitive DNA sequences in lizards: Molecular cytogenetic characterization of a novel family of satellite DNA sequences isolated from the water monitor lizard (Varanus salvator macromaculatus, Platynota). J. Hered. 2013, 104, 798–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Smith, J.M. Analysing the mosaic structure of genes. J. Mol. Evol. 1992, 35, 126–129. [Google Scholar]
- Sawyer, S. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 1989, 6, 526–538. [Google Scholar] [PubMed] [Green Version]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef] [PubMed]
- Ujvari, B.; Dowton, M.; Madsen, T. Mitochondrial DNA recombination in a free-ranging Australian lizard. Biol. Lett. 2007, 3, 189–192. [Google Scholar] [CrossRef]
- Srikulnath, K.; Thongpan, A.; Suputtitada, S.; Apisitwanich, S. New haplotype of the complete mitochondrial genome of Crocodylus siamensis and its species-specific DNA markers: Distinguishing C. siamensis from C. porosus in Thailand. Mol. Biol. Rep. 2012, 39, 4709–4717. [Google Scholar] [CrossRef] [PubMed]
- Laopichienpong, N.; Muangmai, N.; Supikamolseni, A.; Twilprawat, P.; Chanhome, L.; Suntrarachun, S.; Peyachoknagul, S.; Srikulnath, K. Assessment of snake DNA barcodes based on mitochondrial COI and Cytb genes revealed multiple putative cryptic species in Thailand. Gene 2016, 594, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Laopichienpong, N.; Ahmad, S.F.; Singchat, W.; Suntronpong, A.; Pongsanarm, T.; Jangtarwan, K.; Bulan, J.; Pansrikaew, T.; Panthum, T.; Ariyaraphong, N.; et al. Complete mitochondrial genome of Mekong fighting fish, Betta smaragdina (Teleostei: Osphronemidae). Mitochondrial DNA Part B Resour. 2021, 6, 776–778. [Google Scholar] [CrossRef] [PubMed]
- Lapbenjakul, S.; Thapana, W.; Twilprawat, P.; Muangmai, N.; Kanchanaketu, T.; Temsiripong, Y.; Unajak, S.; Peyachoknagul, S.; Srikulnath, K. High genetic diversity and demographic history of captive Siamese and Saltwater crocodiles suggest the first step toward the establishment of a breeding and reintroduction program in Thailand. PLoS ONE 2017, 12, e0184526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.F.; Laopichienpong, N.; Singchat, W.; Suntronpong, A.; Pongsanarm, T.; Panthum, T.; Ariyaraphong, N.; Bulan, J.; Pansrikaew, T.; Jangtarwan, K.; et al. Next-generation sequencing yields complete mitochondrial genome assembly of peaceful betta fish, Betta imbellis (Teleostei: Osphronemidae). Mitochondrial DNA Part B Resour. 2020, 5, 3856–3858. [Google Scholar] [CrossRef] [PubMed]
- Ariyaraphong, N.; Laopichienpong, N.; Singchat, W.; Panthum, T.; Ahmad, S.F.; Jattawa, D.; Duengkae, P.; Muangmai, N.; Suwanasopee, T.; Koonawootrittriron, S.; et al. High-level gene flow restricts genetic differentiation in dairy cattle populations in Thailand: Insights from large-scale mt D-Loop sequencing. Animals 2021, 11, 1680. [Google Scholar] [CrossRef]
- Ariyaraphong, N.; Pansrikaew, T.; Jangtarwan, K.; Thintip, J.; Singchat, W.; Laopichienpong, N.; Pongsanarm, T.; Panthum, T.; Suntronpong, A.; Ahmad, S.F.; et al. Introduction of wild Chinese gorals into a captive population requires careful genetic breeding plan monitoring for successful long-term conservation. Glob. Ecol. Conserv. 2021, 28, e01675. [Google Scholar] [CrossRef]
- Fonseca, M.M.; Harris, D.J.; Posada, D. The inversion of the control region in three mitogenomes provides further evidence for an asymmetric model of vertebrate mtDNA replication. PLoS ONE 2014, 9, e106654. [Google Scholar]
- Eberhard, J.R.; Wright, T.F.; Bermingham, E. Duplication and concerted evolution of the mitochondrial control region in the parrot genus Amazona. Mol. Biol. Evol. 2001, 18, 1330–1342. [Google Scholar] [CrossRef] [Green Version]
- Bronstein, O.; Kroh, A.; Haring, E. Mind the gap! The mitochondrial control region and its power as a phylogenetic marker in echinoids. BMC Evol. Biol. 2018, 18, 80. [Google Scholar] [CrossRef]
- Abbott, C.L.; Double, M.C.; Trueman, J.W.; Robinson, A.; Cockburn, A. An unusual source of apparent mitochondrial heteroplasmy: Duplicate mitochondrial control regions in Thalassarche albatrosses. Mol. Ecol. 2005, 14, 3605–3613. [Google Scholar] [CrossRef]
- Morris-Pocock, J.A.; Taylor, S.A.; Birt, T.P.; Friesen, V.L. Concerted evolution of duplicated mitochondrial control regions in three related seabird species. BMC Evol. Biol. 2010, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Hoarau, G.; Holla, S.; Lescasse, R.; Stam, W.T.; Olsen, J.L. Heteroplasmy and evidence for recombination in the mitochondrial control region of the flatfish Platichthys flesus. Mol. Biol. Evol. 2002, 19, 2261–2264. [Google Scholar] [CrossRef] [Green Version]
- Tsaousis, A.D.; Martin, D.P.; Ladoukakis, E.D.; Posada, D.; Zouros, E. Widespread recombination in published animal mtDNA sequences. Mol. Biol. Evol. 2005, 22, 925–933. [Google Scholar] [CrossRef] [PubMed]




| Region | Within Group | Between Group |
|---|---|---|
| p-Distance | p-Distance | |
| CR1 | 0.39 ± 0.11 | - |
| CR2 | 0.37 ± 0.10 | - |
| CSB_CR1 | 0.40 ± 0.18 | - |
| CSB_CR2 | 0.15 ± 0.09 | - |
| TAS_CR1 | 011 ± 0.11 | - |
| TAS_CR2 | 0.21 ± 0.08 | - |
| CR1 * | 10.83 ± 0.58 | 25.11 ± 5.50 |
| CR2 * | 17.08 ± 0.45 | 38.88 ± 1.54 |
| CSB_CR1 * | 11.23 ± 1.96 | 34.70 ± 6.40 |
| CSB_CR2 * | 14.33 ± 1.88 | 32.49 ± 6.00 |
| TAS_CR1 * | 13.78 ± 0.17 | 41.10 ± 5.50 |
| TAS_CR2 * | 17.08 ± 0.45 | 38.88 ± 1.55 |
| Region | Number of OUT a | Iss b | Iss.cSym c | Df d | p-Value e | Iss.cAsym f | df | p-Value |
|---|---|---|---|---|---|---|---|---|
| CR1 | 4 | 0.028 | 0.805 | 662 | <0.00001 | 0.774 | 662 | 0.0000 |
| 8 | 0.033 | 0.766 | 662 | <0.00001 | 0.656 | 662 | 0.0000 | |
| 16 | 0.039 | 0.745 | 662 | <0.00001 | 0.535 | 662 | 0.0000 | |
| 32 | 0.046 | 0.719 | 662 | <0.00001 | 0.393 | 662 | 0.0000 | |
| CR2 | 4 | 0.007 | 0.815 | 866 | <0.00001 | 0.784 | 866 | 0.0000 |
| 8 | 0.008 | 0.780 | 866 | <0.00001 | 0.673 | 866 | 0.0000 | |
| 16 | 0.008 | 0.763 | 866 | <0.00001 | 0.560 | 866 | 0.0000 | |
| 32 | 0.009 | 0.738 | 866 | <0.00001 | 0.424 | 866 | 0.0000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thapana, W.; Ariyaraphong, N.; Wongtienchai, P.; Laopichienpong, N.; Singchat, W.; Panthum, T.; Ahmad, S.F.; Kraichak, E.; Muangmai, N.; Duengkae, P.; et al. Concerted and Independent Evolution of Control Regions 1 and 2 of Water Monitor Lizards (Varanus salvator macromaculatus) and Different Phylogenetic Informative Markers. Animals 2022, 12, 148. https://doi.org/10.3390/ani12020148
Thapana W, Ariyaraphong N, Wongtienchai P, Laopichienpong N, Singchat W, Panthum T, Ahmad SF, Kraichak E, Muangmai N, Duengkae P, et al. Concerted and Independent Evolution of Control Regions 1 and 2 of Water Monitor Lizards (Varanus salvator macromaculatus) and Different Phylogenetic Informative Markers. Animals. 2022; 12(2):148. https://doi.org/10.3390/ani12020148
Chicago/Turabian StyleThapana, Watcharaporn, Nattakan Ariyaraphong, Parinya Wongtienchai, Nararat Laopichienpong, Worapong Singchat, Thitipong Panthum, Syed Farhan Ahmad, Ekaphan Kraichak, Narongrit Muangmai, Prateep Duengkae, and et al. 2022. "Concerted and Independent Evolution of Control Regions 1 and 2 of Water Monitor Lizards (Varanus salvator macromaculatus) and Different Phylogenetic Informative Markers" Animals 12, no. 2: 148. https://doi.org/10.3390/ani12020148