
Mutation Signatures and In Silico Docking of Novel SARS-CoV-2 Variants of Concern

Abstract
:1. Introduction
2. Material and Methods
2.1. Dataset Collection and Mutation Comparison
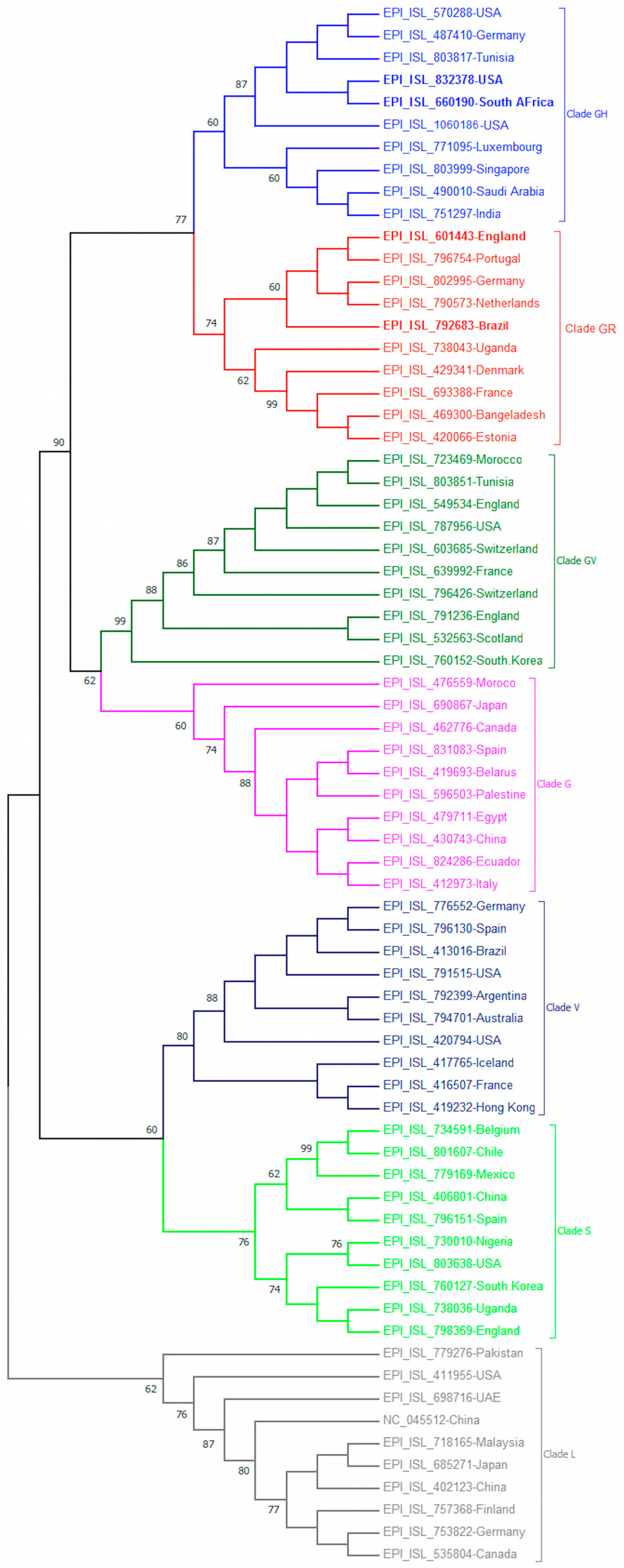
2.2. Phylogenetic Tree Construction
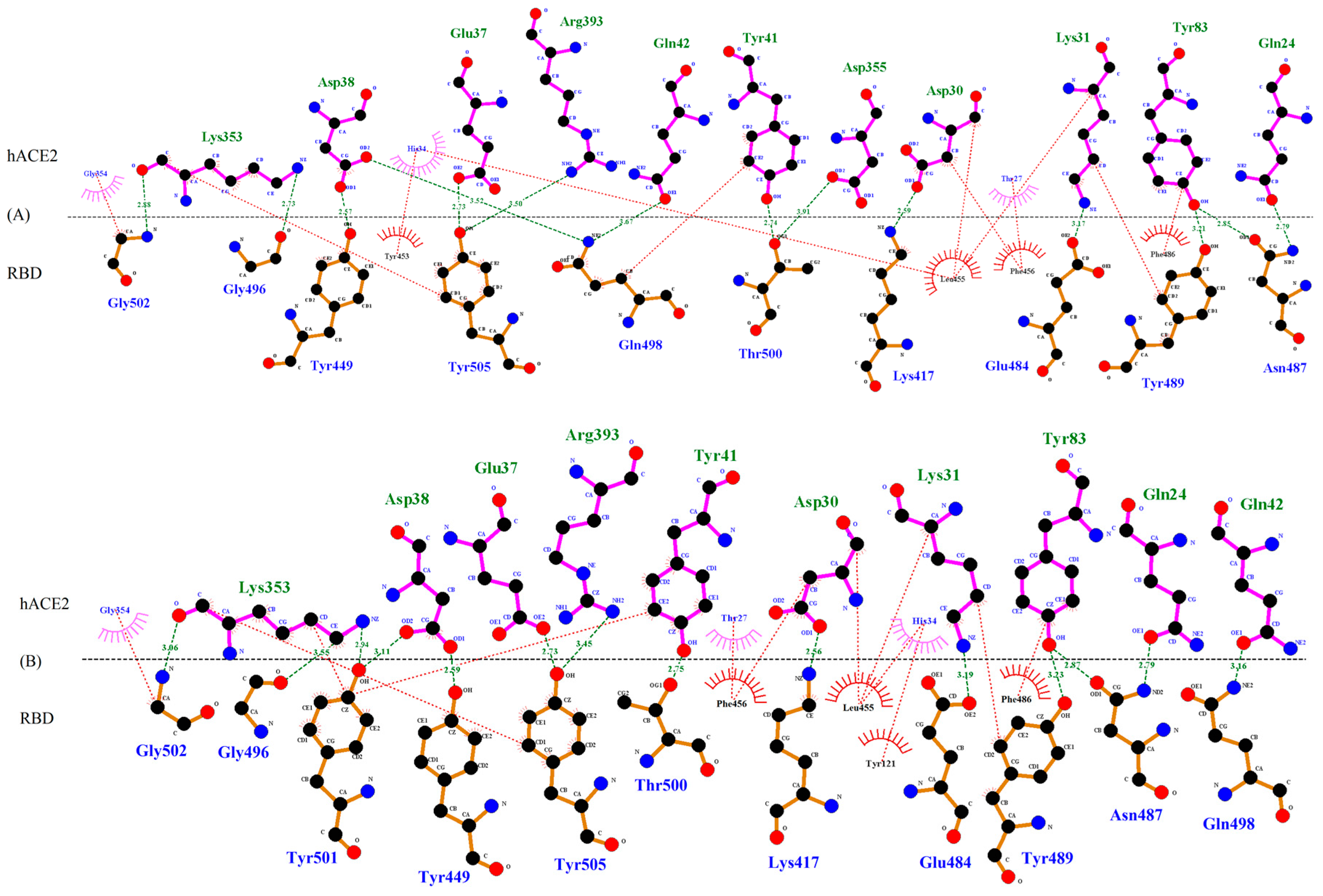
2.3. In Silico Modeling of Molecular Interaction in RBD-hACE2 Complex
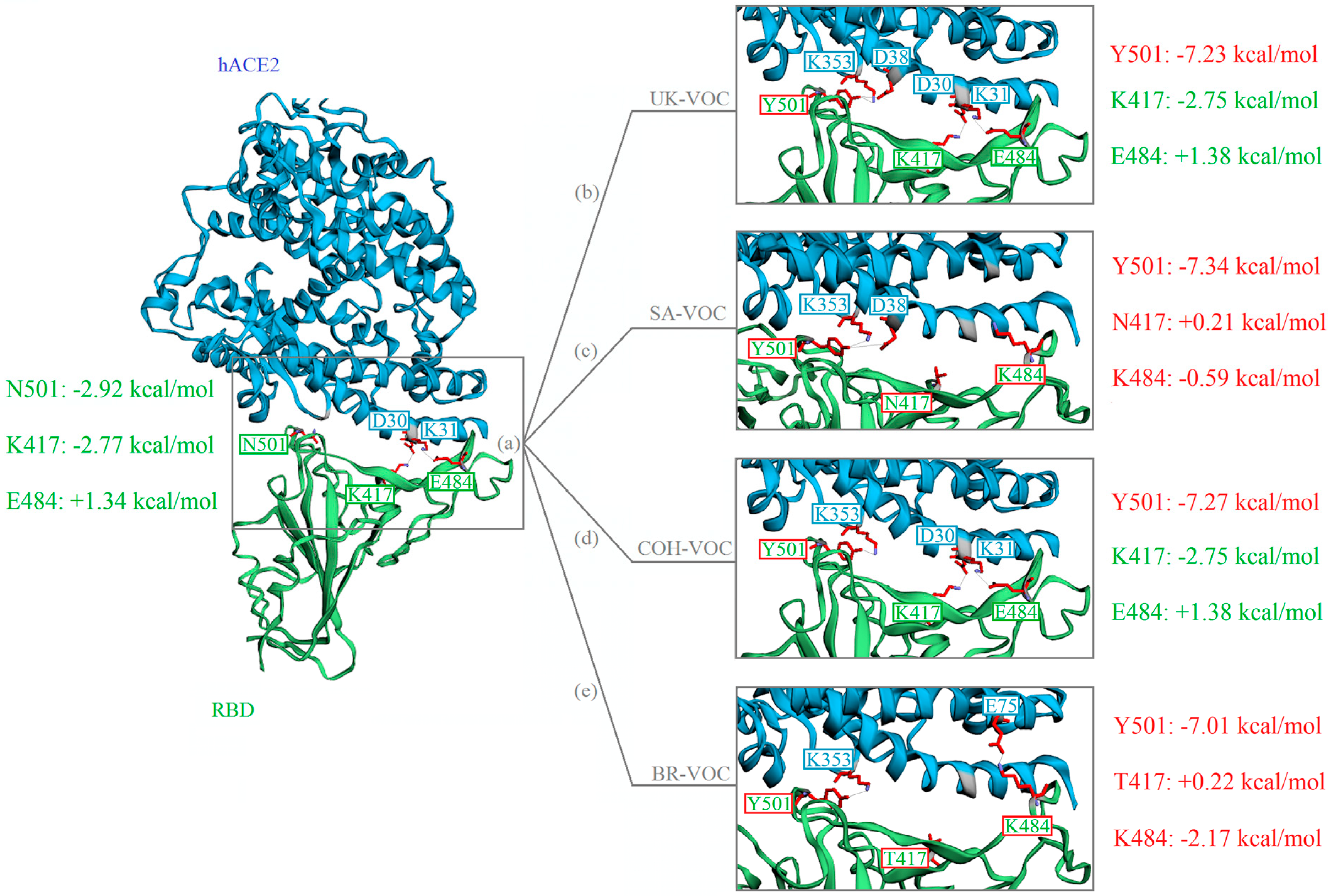
2.4. Molecular Architecture and In Silico FEB of RBD-hACE2
3. Results
3.1. Nonsynonymous Mutations of Novel SARS-CoV-2 Variants
3.2. Phylogeny of Novel SARS-CoV-2 Variants
3.3. Molecular Interactions Between hACE2 and WT or VOC RBD
3.4. FEB of RBD-hACE2 Docked Complexes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Domingo, E. Mechanisms of viral emergence. Veter. Res. 2010, 41, 38. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Alouane, T.; Laamarti, M.; Essabbar, A.; Hakmi, M.; Bouricha, E.M.; Chemao-Elfihri, M.; Kartti, S.; Boumajdi, N.; Bendani, H.; Laamarti, R. Genomic diversity and hotspot muta-tions in 30,983 SARS-CoV-2 genomes: Moving toward a universal vaccine for the “con-fined virus”? Pathogens 2020, 9, 829. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist ge-nomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and genomic distribution of SARS-CoV-2 muta-tions. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020, 592, 116–121. [Google Scholar] [CrossRef]
- Graham, R.L.; Becker, M.M.; Eckerle, L.D.; Bolles, M.; Denison, M.R.; Baric, R.S. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 2012, 18, 1820–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.C.; Blanc, H.; Vignuzzi, M.; Denison, M.R. Coronaviruses Lacking Exoribonuclease Activity Are Susceptible to Lethal Mutagenesis: Evidence for Proofreading and Potential Therapeutics. PLOS Pathog. 2013, 9, e1003565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, P. Rampant C→U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses: Causes and consequences for their short-and long-term evolutionary tra-jectories. Msphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Avenarius, M.R.; Kubatko, L.; Hunt, M.; Pan, X.; Ru, P.; Garee, J.; Thomas, K.; Mohler, P.; Pancholi, P. Distinct Patterns of Emergence of SARS-CoV-2 Spike Variants in-cluding N501Y in Clinical Samples in Columbus Ohio. bioRxiv 2021. [Google Scholar] [CrossRef]
- Resende, P.C.; Bezerra, J.F.; de Vasconcelos, R.H.T.; Arantes, I.; Appolinario, L.; Mendon-ça, A.C.; Paixao, A.C.; Rodrigues, A.C.D.; Silva, T.; Rocha, A.S. Spike E484K mutation in the first SARS-CoV-2 reinfection case confirmed in Brazil. Genom. Epidem. 2021, 10, 2021. [Google Scholar]
- Zou, J.; Yin, J.; Fang, L.; Yang, M.; Wang, T.; Wu, W.; Bellucci, M.A.; Zhang, P. Computa-tional Prediction of Mutational Effects on SARS-CoV-2 Binding by Relative Free Energy Calculations. J. Chem. Inf. Model. 2020, 60, 5794–5802. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.K.; Maurya, A.K.; Mishra, N.; Siddique, H.R. Virtual screening, ADME/T, and binding free energy analysis of anti-viral, anti-protease, and anti-infectious compounds against NSP10/NSP16 methyltransferase and main protease of SARS CoV-2. J. Recept. Signal. Transduct. 2020, 40, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Jakhmola, S.; Indari, O.; Kashyap, D.; Varshney, N.; Das, A.; Manivannan, E.; Jha, H.C. Mutational analysis of structural proteins of SARS-CoV-2. Heliyon 2021, 7, e06572. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.A.; Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997; Volume 12. [Google Scholar]
- Marqusee, S.; Baldwin, R.L. Helix stabilization by Glu-... Lys+ salt bridges in short pep-tides of de novo design. Proc. Nat. Acad. Sci. USA 1987, 84, 8898–8902. [Google Scholar] [CrossRef] [Green Version]
- Thanh Le, T.; Andreadakis, Z.; Kumar, A.; Gómez Román, R.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef]
- Shahhosseini, N.; Frederick, C.; Letourneau-Montminy, M.-P.; Marie-Odile, B.-B.; Kobinger, G.P.; Wong, G. Computational genomics of Torque teno sus virus and Porcine circovirus in swine samples from Canada. Res. Veter. Sci. 2021, 134, 171–180. [Google Scholar] [CrossRef]
- Shahhosseini, N.; Moosa-Kazemi, S.H.; Sedaghat, M.M.; Wong, G.; Chinikar, S.; Hajivand, Z.; Mokhayeri, H.; Nowotny, N.; Kayedi, M.H. Autochthonous Transmission of West Nile Virus by a New Vector in Iran, Vector-Host Interaction Modeling and Virulence Gene Determinants. Viruses 2020, 12, 1449. [Google Scholar] [CrossRef]
- Joosten, R.P.; Beek, T.A.H.T.; Krieger, E.; Hekkelman, M.L.; Hooft, R.W.W.; Schneider, R.; Sander, C.; Vriend, G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2010, 39, D411–D419. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein–protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. The accuracy of binding free energy calculations based on molec-ular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein–protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- Shahhosseini, N.; Wong, G.; Kobinger, G.P.; Chinikar, S. SARS-CoV-2 spillover trans-mission due to recombination event. Gene Rep. 2021, 23, 101045. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, A.; Kandeil, A.; Shehata, M.; Shesheny, R.E.; Samy, A.M.; Kayali, G.; Ali, M.A. Middle east respiratory syndrome coronavirus (mers-cov): State of the science. Microorganisms 2020, 8, 991. [Google Scholar] [CrossRef] [PubMed]
- Zhukova, A.; Blassel, L.; Lemoine, F.; Morel, M.; Voznica, J.; Gascuel, O. Origin, evolution and global spread of SARS-CoV-Comptes Rendus. Biologies 2020. [Google Scholar] [CrossRef]
- Hayashi, T.; Yaegashi, N.; Konishi, I. Effect of RBD mutation (Y453F) in spike glycoprotein of SARS-CoV-2 on neutralizing antibody affinity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA 2021, 325, 529. [Google Scholar] [CrossRef]
- Davies, N.G.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.; Pearson, C.A.; Russell, T.W.; Tully, D.C.; Abbott, S.; Gimma, A. Estimated transmissibility and severity of novel SARS-CoV-2 Variant of Concern 202012/01 in England. medRxiv 2020. [Google Scholar] [CrossRef]
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A. Preliminary ge-nomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Genom. Epidemiol. 2020, 1–5. [Google Scholar]
- Cohen, J. Vaccine Designers Take First Shots at COVID-19; American Association for the Advancement of Science (AAAS): Washington, DC, USA, 2020. [Google Scholar]
- Xie, X.; Muruato, A.; Lokugamage, K.G.; Narayanan, K.; Zhang, X.; Zou, J.; Liu, J.; Schindewolf, C.; Bopp, N.E.; Aguilar, P.V. An infectious cDNA clone of SARS-CoV-Cell. Host Microb. 2020, 27, 841–848.e3. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. Capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking. Phys. Chem. Chem. Phys. 2016, 18, 22129–22139. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, M.; Brooks, B.R.; Klauda, J.B. Critical Sequence Hotspots for Binding of Novel Coronavirus to Angiotensin Converter Enzyme as Evaluated by Molecular Simulations. J. Phys. Chem. B 2020, 124, 10034–10047. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.; Carter, H.; Zanetti, M. Potential global impact of the N501Y mutation on MHC-II presentation and immune escape. bioRxiv 2021. [Google Scholar] [CrossRef]
- Villoutreix, B.; Calvez, V.; Marcelin, A.-G.; Khatib, A.-M. In Silico Investigation of the New UK (B.1.1.7) and South African (501Y.V2) SARS-CoV-2 Variants with a Focus at the ACE2–Spike RBD Interface. Int. J. Mol. Sci. 2021, 22, 1695. [Google Scholar] [CrossRef]
- Nelson, G.; Buzko, O.; Spilman, P.R.; Niazi, K.; Rabizadeh, S.; Soon-Shiong, P.R. Molecular dynamic simulation reveals E484K mutation enhances spike RBD-ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y. V2 variant) induces conforma-tional change greater than N501Y mutant alone, potentially resulting in an escape mutant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhang, W.; Davis, B.D.; Chen, S.S.; Martinez, J.M.S.; Plummer, J.T.; Vail, E. Emergence of a Novel SARS-CoV-2 Variant in Southern California. JAMA 2021, 325, 1324–1326. [Google Scholar] [CrossRef] [PubMed]
- Dagotto, G.; Yu, J.; Barouch, D.H. Approaches and challenges in SARS-CoV-2 vaccine de-velopment. Cell Host Microb. 2020, 28, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.; Chen, R.; Xie, X.; Case, J.; Zhang, X.; VanBlargan, L.; Liu, Y.; Liu, J.; Errico, J.; Winkler, E. SARS-CoV-2 variants show resistance to neutralization by many monoclonal and serum-derived polyclonal antibodies. Res. Square 2021. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequences | Ref. Seq. | UK-VOC | SA-VOC | COH-VOC | BR-VOC | |
|---|---|---|---|---|---|---|---|
| aa Position | |||||||
| ORF1ab | NSP2-4 | R | R | C | R | R | |
| NSP2-85 | T | T | T | I | T | ||
| NSP3-181 | T | T | T | I | T | ||
| NSP3-183 | T | I | T | T | T | ||
| NSP3-256 | A | A | A | V | A | ||
| NSP3-370 | S | S | S | S | L | ||
| NSP3-837 | K | K | N | K | K | ||
| NSP3-890 | A | D | A | A | A | ||
| NSP3-977 | K | K | K | K | Q | ||
| NSP3-1412 | I | T | I | I | I | ||
| NSP5-89 | L | L | L | F | L | ||
| NSP5-108 | P | P | P | S | P | ||
| NSP6-106-108 | SGF | DEL | SGF | SGF | DEL | ||
| NSP6-258 | G | G | G | E | G | ||
| NSP12-323 | P | L | L | L | L | ||
| NSP13-168 | E | E | D | E | E | ||
| NSP13-341 | E | E | E | E | D | ||
| NSP14-28 | S | S | C | S | S | ||
| NSP14-129 | N | N | N | D | N | ||
| NSP15-205 | P | P | L | P | P | ||
| NSP16-216 | R | R | R | C | R | ||
| S | S-18 | L | L | L | L | F | |
| S-20 | T | T | T | T | N | ||
| S-26 | P | P | P | P | S | ||
| S-69-70 | HV | DEL | HV | HV | HV | ||
| S-80 | D | D | A | D | D | ||
| S-138 | D | D | D | D | Y | ||
| S-144 | Y | DEL | Y | Y | |||
| S-190 | R | R | R | R | S | ||
| S-215 | D | D | G | D | D | ||
| S-241-243 | LLA | LLA | DEL | LLA | LLA | ||
| S-RBD | S-417 | K | K | N | K | T | |
| S-484 | E | E | K | E | K | ||
| S-501 | N | Y | Y | Y | Y | ||
| S-570 | A | D | A | A | A | ||
| S-614 | D | G | G | G | G | ||
| S-655 | H | H | H | H | Y | ||
| S-OGD | S-681 | P | H | P | P | P | |
| S-701 | A | A | V | A | A | ||
| S-716 | T | I | T | T | T | ||
| S-982 | S | A | S | S | S | ||
| S-1027 | T | T | T | T | I | ||
| S-1118 | D | H | D | D | D | ||
| S-1176 | V | V | V | V | F | ||
| ORF3 | NS3-57 | Q | Q | H | H | Q | |
| NS3-171 | S | S | L | S | S | ||
| NS3-172 | G | G | G | V | G | ||
| NS3-253 | S | S | S | S | P | ||
| E | E-71 | P | P | L | P | P | |
| M | - | - | - | - | - | - | |
| ORF6 | - | - | - | - | - | - | |
| ORF7a | NS7a-120 | T | T | T | I | T | |
| ORF7b | - | - | - | - | - | - | |
| ORF8 | NS8-24 | S | S | S | L | S | |
| NS8-52 | R | I | R | R | R | ||
| NS8-73 | Y | C | Y | Y | Y | ||
| NS8-92 | E | E | E | E | K | ||
| N | N-3 | D | L | D | D | D | |
| N-67 | P | P | P | S | R | ||
| N-80 | P | P | P | P | R | ||
| N-199 | P | P | P | L | P | ||
| N-203 | R | K | R | R | K | ||
| N-204 | G | R | G | G | R | ||
| N-205 | T | T | I | T | T | ||
| N-235 | S | F | S | S | S | ||
| ORF10 | - | - | - | - | - | - | |
| Total number | Mutation/deletion | NA | 20 | 18 | 17 | 23 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahhosseini, N.; Babuadze, G.; Wong, G.; Kobinger, G.P. Mutation Signatures and In Silico Docking of Novel SARS-CoV-2 Variants of Concern. Microorganisms 2021, 9, 926. https://doi.org/10.3390/microorganisms9050926
Shahhosseini N, Babuadze G, Wong G, Kobinger GP. Mutation Signatures and In Silico Docking of Novel SARS-CoV-2 Variants of Concern. Microorganisms. 2021; 9(5):926. https://doi.org/10.3390/microorganisms9050926
Chicago/Turabian StyleShahhosseini, Nariman, George (Giorgi) Babuadze, Gary Wong, and Gary P. Kobinger. 2021. "Mutation Signatures and In Silico Docking of Novel SARS-CoV-2 Variants of Concern" Microorganisms 9, no. 5: 926. https://doi.org/10.3390/microorganisms9050926