
Information Scale Correction for Varying Length Amplicons Improves Eukaryotic Microbiome Data Integration

Abstract
:1. Introduction
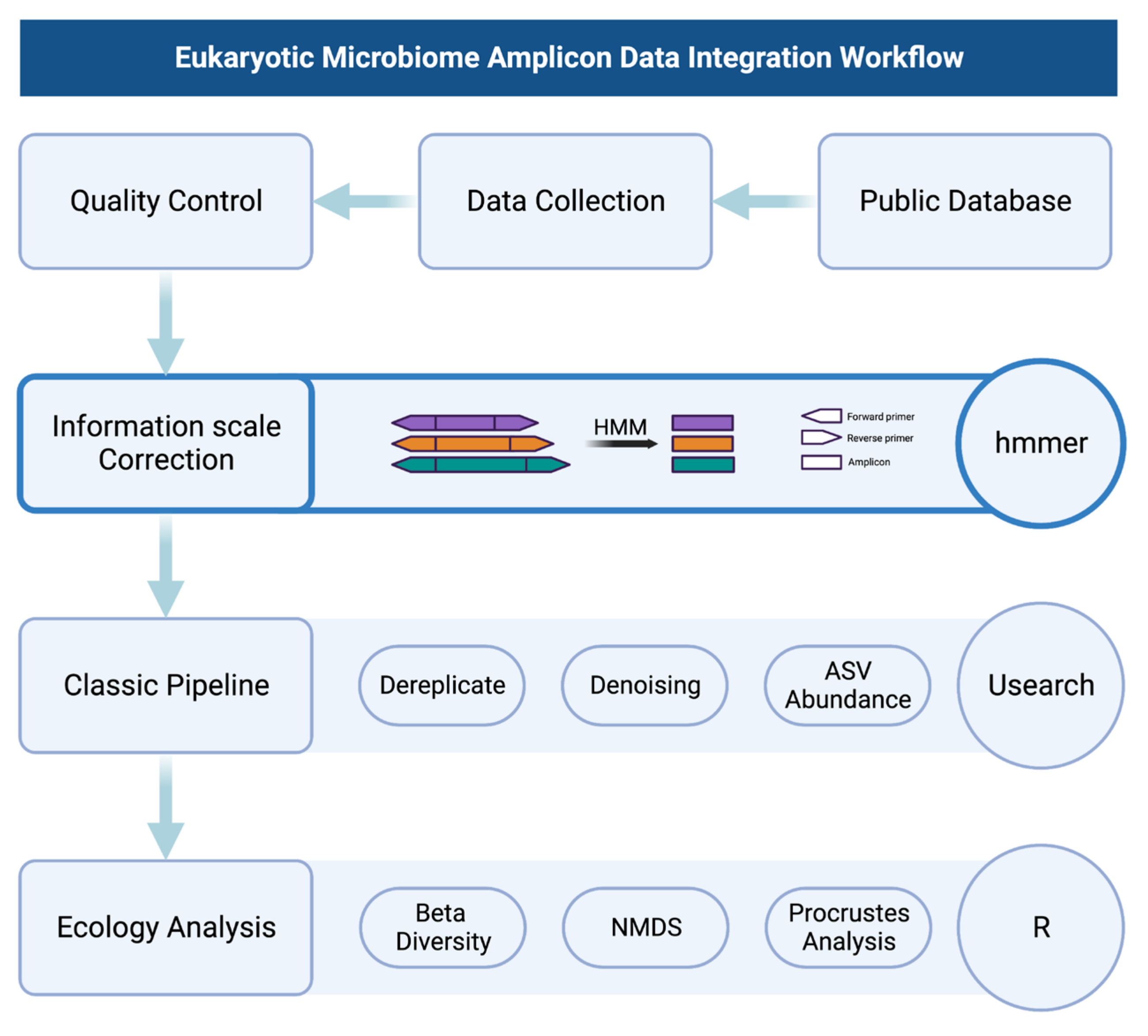
2. Materials and Methods
2.1. Data Collection
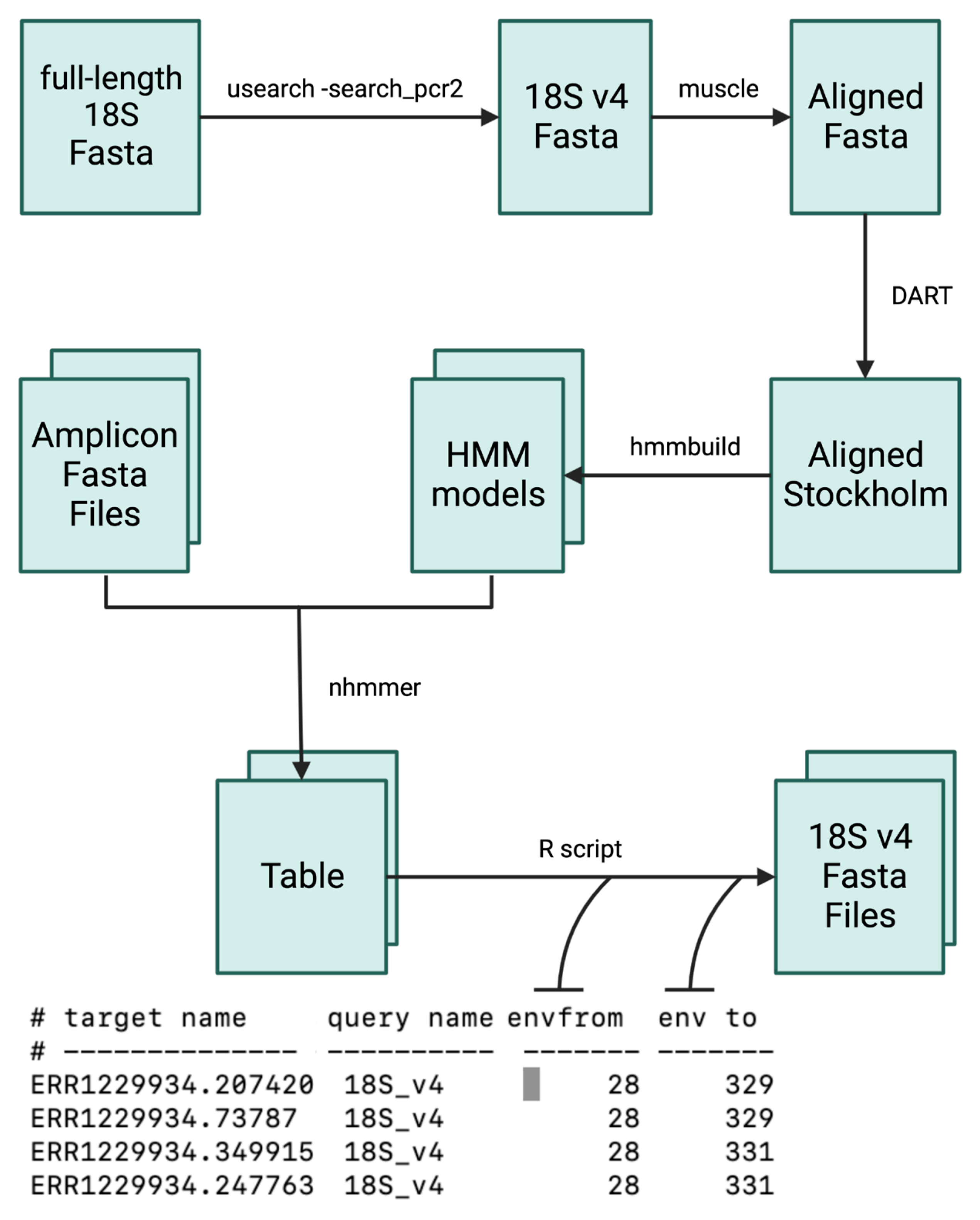
2.2. Information Scale Correction
2.3. Bioinformatics Analysis
2.4. Statistical Analysis
3. Result
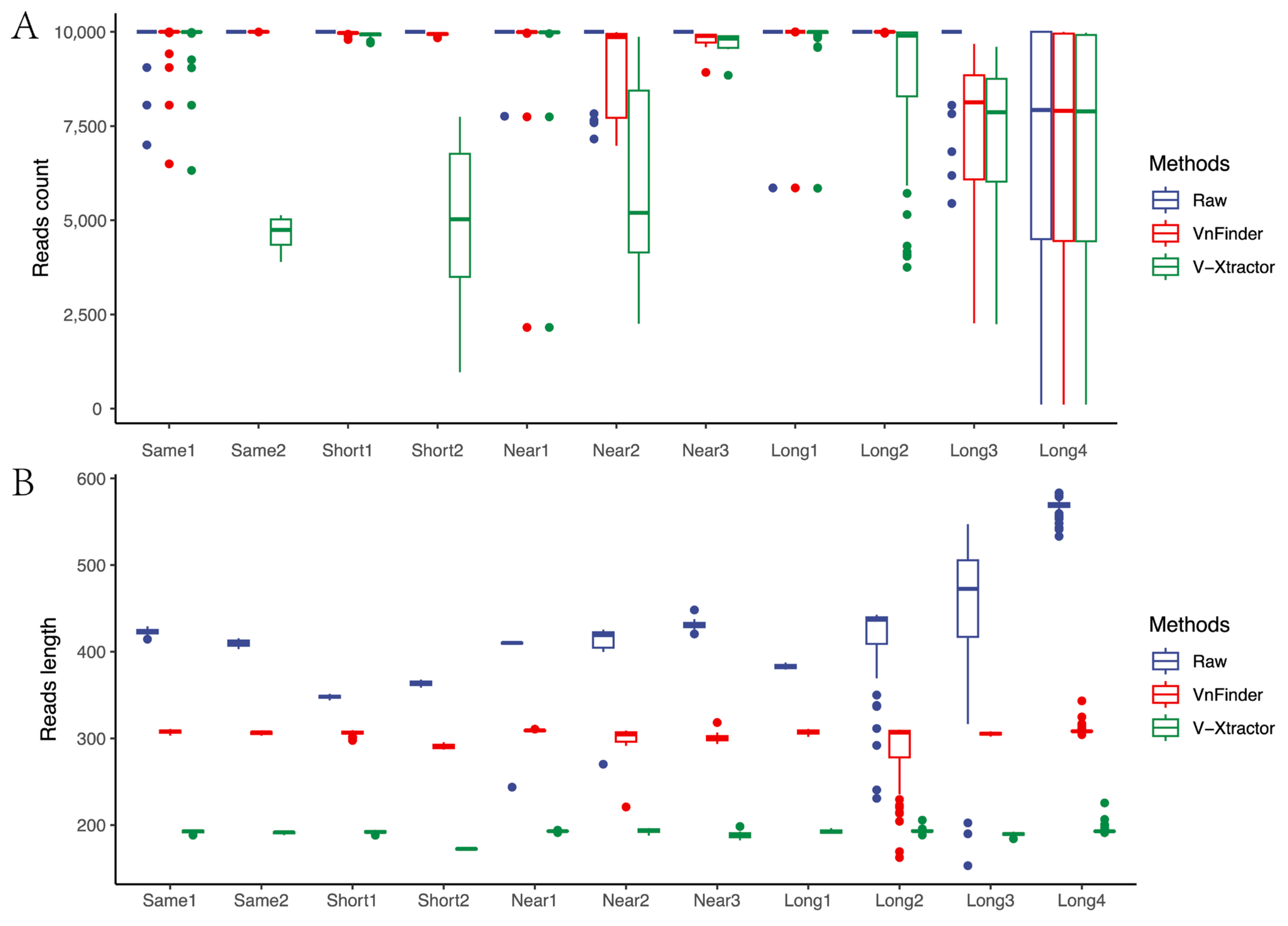
3.1. Correction Effect Comparison
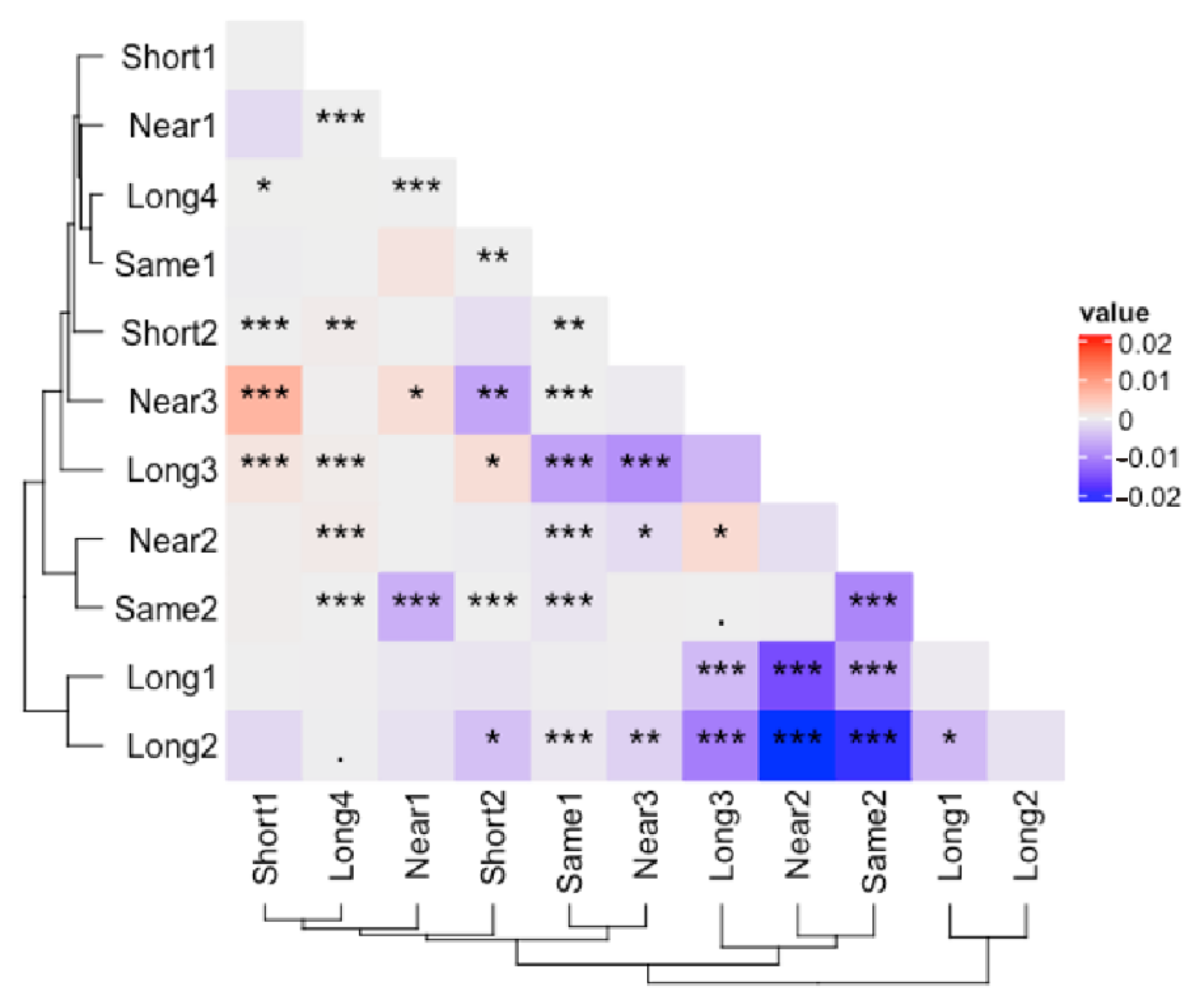
3.2. Corrected and Non-Differential Effects
3.3. Correction Effect in Relation to Amplicon Region
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tara Ocean Foundation; Abreu, A.; Bourgois, E.; Gristwood, A.; Troublé, R.; Tara Oceans; Acinas, S.G.; Bork, P.; Boss, E.; Bowler, C.; et al. Priorities for Ocean Microbiome Research. Nat. Microbiol. 2022, 7, 937–947. [Google Scholar] [CrossRef]
- Cordier, T.; Angeles, I.B.; Henry, N.; Lejzerowicz, F.; Berney, C.; Morard, R.; Brandt, A.; Cambon-Bonavita, M.-A.; Guidi, L.; Lombard, F.; et al. Patterns of Eukaryotic Diversity from the Surface to the Deep-Ocean Sediment. Sci. Adv. 2022, 8, eabj9309. [Google Scholar] [CrossRef]
- Villarino, E.; Watson, J.R.; Jönsson, B.; Gasol, J.M.; Salazar, G.; Acinas, S.G.; Estrada, M.; Massana, R.; Logares, R.; Giner, C.R.; et al. Large-Scale Ocean Connectivity and Planktonic Body Size. Nat. Commun. 2018, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Jamy, M.; Foster, R.; Barbera, P.; Czech, L.; Kozlov, A.; Stamatakis, A.; Bending, G.; Hilton, S.; Bass, D.; Burki, F. Long-read Metabarcoding of the Eukaryotic RDNA Operon to Phylogenetically and Taxonomically Resolve Environmental Diversity. Mol. Ecol. Resour. 2020, 20, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Schlaeppi, K.; Bender, S.F.; Mascher, F.; Russo, G.; Patrignani, A.; Camenzind, T.; Hempel, S.; Rillig, M.C.; Heijden, M.G.A. High-resolution Community Profiling of Arbuscular Mycorrhizal Fungi. New Phytol. 2016, 212, 780–791. [Google Scholar] [CrossRef]
- McDonald, D.; Jiang, Y.; Balaban, M.; Cantrell, K.; Zhu, Q.; Gonzalez, A.; Morton, J.T.; Nicolaou, G.; Parks, D.H.; Karst, S.; et al. Greengenes2 Enables a Shared Data Universe for Microbiome Studies. bioRxiv 2022. [Google Scholar] [CrossRef]
- Franzén, O.; Hu, J.; Bao, X.; Itzkowitz, S.H.; Peter, I.; Bashir, A. Improved OTU-Picking Using Long-Read 16S RRNA Gene Amplicon Sequencing and Generic Hierarchical Clustering. Microbiome 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, L.J.; Soubrier, J.; Weyrich, L.S.; Cooper, A. Environmental Metabarcodes for Insects: In Silico PCR Reveals Potential for Taxonomic Bias. Mol. Ecol. Resour. 2014, 14, 1160–1170. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Pereira, M.; Zha, Y.; Seifert, M.; Kaldhusdal, V.; Boulund, F.; Krog, M.C.; Bashir, Z.; Hamsten, M.; Fransson, E.; et al. Assessment of In Vitro and In Silico Protocols for Sequence-Based Characterization of the Human Vaginal Microbiome. mSphere 2020, 5, e00448-20. [Google Scholar] [CrossRef]
- Brasseur, M.V.; Astrin, J.J.; Geiger, M.F.; Mayer, C. MitoGeneExtractor: Efficient Extraction of Mitochondrial Genes from Next-generation Sequencing Libraries. Methods Ecol. Evol. 2023, 1–8. [Google Scholar] [CrossRef]
- Allio, R.; Schomaker-Bastos, A.; Romiguier, J.; Prosdocimi, F.; Nabholz, B.; Delsuc, F. MitoFinder: Efficient Automated Large-scale Extraction of Mitogenomic Data in Target Enrichment Phylogenomics. Mol. Ecol. Resour. 2020, 20, 892–905. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson-Palme, J.; Ryberg, M.; Hartmann, M.; Branco, S.; Wang, Z.; Godhe, A.; De Wit, P.; Sánchez-García, M.; Ebersberger, I.; de Sousa, F.; et al. Improved Software Detection and Extraction of ITS1 and ITS2 from Ribosomal ITS Sequences of Fungi and Other Eukaryotes for Analysis of Environmental Sequencing Data. Methods Ecol. Evol. 2013, 4, 914–919. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Hartmann, M.; Eriksson, K.M.; Pal, C.; Thorell, K.; Larsson, D.G.J.; Nilsson, R.H. metaxa 2: Improved Identification and Taxonomic Classification of Small and Large Subunit RRNA in Metagenomic Data. Mol. Ecol. Resour. 2015, 15, 1403–1414. [Google Scholar] [CrossRef]
- Hartmann, M.; Howes, C.G.; Abarenkov, K.; Mohn, W.W.; Nilsson, R.H. V-Xtractor: An Open-Source, High-Throughput Software Tool to Identify and Extract Hypervariable Regions of Small Subunit (16S/18S) Ribosomal RNA Gene Sequences. J. Microbiol. Methods 2010, 83, 250–253. [Google Scholar] [CrossRef]
- Büttner, M.; Miao, Z.; Wolf, F.A.; Teichmann, S.A.; Theis, F.J. A Test Metric for Assessing Single-Cell RNA-Seq Batch Correction. Nat. Methods 2019, 16, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karst, S.M.; Ziels, R.M.; Kirkegaard, R.H.; Sørensen, E.A.; McDonald, D.; Zhu, Q.; Knight, R.; Albertsen, M. High-Accuracy Long-Read Amplicon Sequences Using Unique Molecular Identifiers with Nanopore or PacBio Sequencing. Nat. Methods 2021, 18, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Song, X.; Zhang, C.-Y.; Chen, G.-F.; Lao, Y.-M.; Jin, H.; Cai, Z.-H. Distribution Patterns of Microbial Community Structure Along a 7000-Mile Latitudinal Transect from the Mediterranean Sea Across the Atlantic Ocean to the Brazilian Coastal Sea. Microb. Ecol. 2018, 76, 592–609. [Google Scholar] [CrossRef]
- Vaulot, D.; Geisen, S.; Mahé, F.; Bass, D. Pr2-primers: An 18S RRNA Primer Database for Protists. Mol. Ecol. Resour. 2022, 22, 168–179. [Google Scholar] [CrossRef]
- Brown, M.S.; Bowman, J.S.; Lin, Y.; Feehan, C.J.; Moreno, C.M.; Cassar, N.; Marchetti, A.; Schofield, O.M. Low Diversity of a Key Phytoplankton Group along the West Antarctic Peninsula. Limnol. Oceanogr. 2021, 66, 2470–2480. [Google Scholar] [CrossRef]
- Annenkova, N.V.; Giner, C.R.; Logares, R. Tracing the Origin of Planktonic Protists in an Ancient Lake. Microorganisms 2020, 8, 543. [Google Scholar] [CrossRef] [Green Version]
- Enberg, S.; Majaneva, M.; Autio, R.; Blomster, J.; Rintala, J. Phases of Microalgal Succession in Sea Ice and the Water Column in the Baltic Sea from Autumn to Spring. Mar. Ecol. Prog. Ser. 2018, 599, 19–34. [Google Scholar] [CrossRef] [Green Version]
- Fiore-Donno, A.M.; Rixen, C.; Rippin, M.; Glaser, K.; Samolov, E.; Karsten, U.; Becker, B.; Bonkowski, M. New Barcoded Primers for Efficient Retrieval of Cercozoan Sequences in High-Throughput Environmental Diversity Surveys, with Emphasis on Worldwide Biological Soil Crusts. Mol. Ecol. Resour. 2018, 18, 229–239. [Google Scholar] [CrossRef]
- Fadeev, E.; Salter, I.; Schourup-Kristensen, V.; Nöthig, E.-M.; Metfies, K.; Engel, A.; Piontek, J.; Boetius, A.; Bienhold, C. Microbial Communities in the East and West Fram Strait During Sea Ice Melting Season. Front. Mar. Sci. 2018, 5, 429. [Google Scholar] [CrossRef] [Green Version]
- Belevich, T.A.; Ilyash, L.V.; Milyutina, I.A.; Logacheva, M.D.; Goryunov, D.V.; Troitsky, A.V. Photosynthetic Picoeukaryotes in the Land-Fast Ice of the White Sea, Russia. Microb. Ecol. 2018, 75, 582–597. [Google Scholar] [CrossRef] [PubMed]
- Boscaro, V.; Rossi, A.; Vannini, C.; Verni, F.; Fokin, S.I.; Petroni, G. Strengths and Biases of High-Throughput Sequencing Data in the Characterization of Freshwater Ciliate Microbiomes. Microb. Ecol. 2017, 73, 865–875. [Google Scholar] [CrossRef]
- Bradley, I.M.; Pinto, A.J.; Guest, J.S. Design and Evaluation of Illumina MiSeq-Compatible, 18S RRNA Gene-Specific Primers for Improved Characterization of Mixed Phototrophic Communities. Appl. Environ. Microbiol. 2016, 82, 5878–5891. [Google Scholar] [CrossRef] [Green Version]
- Kwong, W.K.; del Campo, J.; Mathur, V.; Vermeij, M.J.A.; Keeling, P.J. A Widespread Coral-Infecting Apicomplexan with Chlorophyll Biosynthesis Genes. Nature 2019, 568, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Geisen, S.; Snoek, L.B.; ten Hooven, F.C.; Duyts, H.; Kostenko, O.; Bloem, J.; Martens, H.; Quist, C.W.; Helder, J.A.; der Putten, W.H. Integrating Quantitative Morphological and Qualitative Molecular Methods to Analyse Soil Nematode Community Responses to Plant Range Expansion. Methods Ecol. Evol. 2018, 9, 1366–1378. [Google Scholar] [CrossRef] [Green Version]
- Venter, P.C.; Nitsche, F.; Domonell, A.; Heger, P.; Arndt, H. The Protistan Microbiome of Grassland Soil: Diversity in the Mesoscale. Protist 2017, 168, 546–564. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle5: High-Accuracy Alignment Ensembles Enable Unbiased Assessments of Sequence Homology and Phylogeny. Nat. Commun. 2022, 13, 6968. [Google Scholar] [CrossRef]
- Wheeler, T.J.; Eddy, S.R. Nhmmer: DNA Homology Search with Profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pages, H.; Aboyoun, P.; Gentleman, R.; DebRoy, S. Biostrings: String objects representing biological sequences, and matching algorithms. R Package Version 2016, 2, 10–18129. [Google Scholar]
- Schloss, P.D. Amplicon Sequence Variants Artificially Split Bacterial Genomes into Separate Clusters. mSphere 2021, 6, e00191-21. [Google Scholar] [CrossRef]
- Edgar, R.C. UNOISE2: Improved Error-Correction for Illumina 16S and ITS Amplicon Sequencing. bioRxiv 2016, 15, 081257. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Pitz, K.J.; Guo, J.; Johnson, S.B.; Campbell, T.L.; Zhang, H.; Vrijenhoek, R.C.; Chavez, F.P.; Geller, J. Zooplankton Biogeographic Boundaries in the California Current System as Determined from Metabarcoding. PLoS ONE 2020, 15, e0235159. [Google Scholar] [CrossRef] [PubMed]
- Harder, C.B.; Rønn, R.; Brejnrod, A.; Bass, D.; Al-Soud, W.A.; Ekelund, F. Local Diversity of Heathland Cercozoa Explored by In-Depth Sequencing. ISME J. 2016, 10, 2488–2497. [Google Scholar] [CrossRef] [Green Version]
- Balzano, S.; Abs, E.; Leterme, S. Protist Diversity along a Salinity Gradient in a Coastal Lagoon. Aquat. Microb. Ecol. 2015, 74, 263–277. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Li, Y.; Lu, Y.; Li, Y.; Yuan, Z.; Dai, M.; Liu, H. Impacts of the Zhe-Min Coastal Current on the Biogeographic Pattern of Microbial Eukaryotic Communities. Prog. Oceanogr. 2020, 183, 102309. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group ID | Forward Primer | Start Position | Reverse Primer | End Position | Sample Size | Source (ID) | Reference |
|---|---|---|---|---|---|---|---|
| Same1 | CCAGCASCYGCGGTAATTCC | 564 | ACTTTCGTTCTTGAT | 980 | 120 | EBI-ENA (PRJNA508517) | [19] |
| Same2 | CCAGCASCYGCGGTAAT | 564 | ACTTTCGTTCTTGATYRA | 980 | 23 | NCBI-SRA (PRJEB24415) | [20] |
| Short1 | CYGCGGTAATTCCAGCTC | 571 | TCYDAGAATTYCACCTCT | 914 | 73 | NCBI-SRA (PRJEB21047) | [21] |
| Short2 | TTAAAAAGCTCGTAGTTG | 616 | AAGAAGACATCCTTGGTG | 963 | 27 | NCBI-SRA (SRR5189947) | [22] |
| Near1 | GCGGTAATTCCAGCTCCAA | 573 | ACTTTCGTTCTTGATYRR | 980 | 33 | EBI-ENA (PRJEB26288) | [23] |
| Near2 | CCAGCASCCGCGGTAATWCC | 564 | AKCCCCYAACTTTCGTTCTTGAT | 988 | 17 | NCBI-SRA (PRJNA368621) | [24] |
| Near3 | CCAGCASCCGCGGTAATWCC | 564 | TCTGRTYGTCTTTGATCCCYTA | 1002 | 12 | EBI-ENA (PRJEB12534) | [25] |
| Long1 | CGGTAAYTCCAGCTCYAV | 574 | CCGTCAATTHCTTYAART | 1149 | 62 | NCBI-SRA (SRP071862) | [26] |
| Long2 | GTGCCAGCAGCCGCG | 561 | TTTAAGTTTCAGCCTTGCG | 1138 | 44 | NCBI-SRA (PRJNA482746) | [27] |
| Long3 | GGCAAGTCTGGTGCCAG | 551 | TCCGTCAATTYCTTTAAGT | 1149 | 36 | EBI-ENA (PRJEB24755) | [28] |
| Long4 | CGGTAATTCCAGCTCCAATAGC | 574 | CACCAACTAAGAACGGCCATGC | 1293 | 150 | NCBI-SRA (SRP101780) | [29] |
| Silva | PR2 | |
|---|---|---|
| Version | 18S v123 | v4.14.0 |
| Total reads | 138,553 | 197,602 |
| Full length (>1600 bp) | 112,110 (80.91%) | 94,283 (47.71%) |
| Partical (<1000 bp) | 293 (0.21%) | 48,002 (24.29%) |
| v4 by VnFinder | 136,228 (98.32%) | 174,646 (88.38%) |
| v4 by V-Xtractor | 132,770 (95.83%) | 151,393 (76.62%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, T.; Zhao, F.; Xu, K. Information Scale Correction for Varying Length Amplicons Improves Eukaryotic Microbiome Data Integration. Microorganisms 2023, 11, 949. https://doi.org/10.3390/microorganisms11040949
Zhou T, Zhao F, Xu K. Information Scale Correction for Varying Length Amplicons Improves Eukaryotic Microbiome Data Integration. Microorganisms. 2023; 11(4):949. https://doi.org/10.3390/microorganisms11040949
Chicago/Turabian StyleZhou, Tong, Feng Zhao, and Kuidong Xu. 2023. "Information Scale Correction for Varying Length Amplicons Improves Eukaryotic Microbiome Data Integration" Microorganisms 11, no. 4: 949. https://doi.org/10.3390/microorganisms11040949