
HIV/Mtb Co-Infection: From the Amplification of Disease Pathogenesis to an “Emerging Syndemic”
 , ,
, ,  , and
, and
Abstract
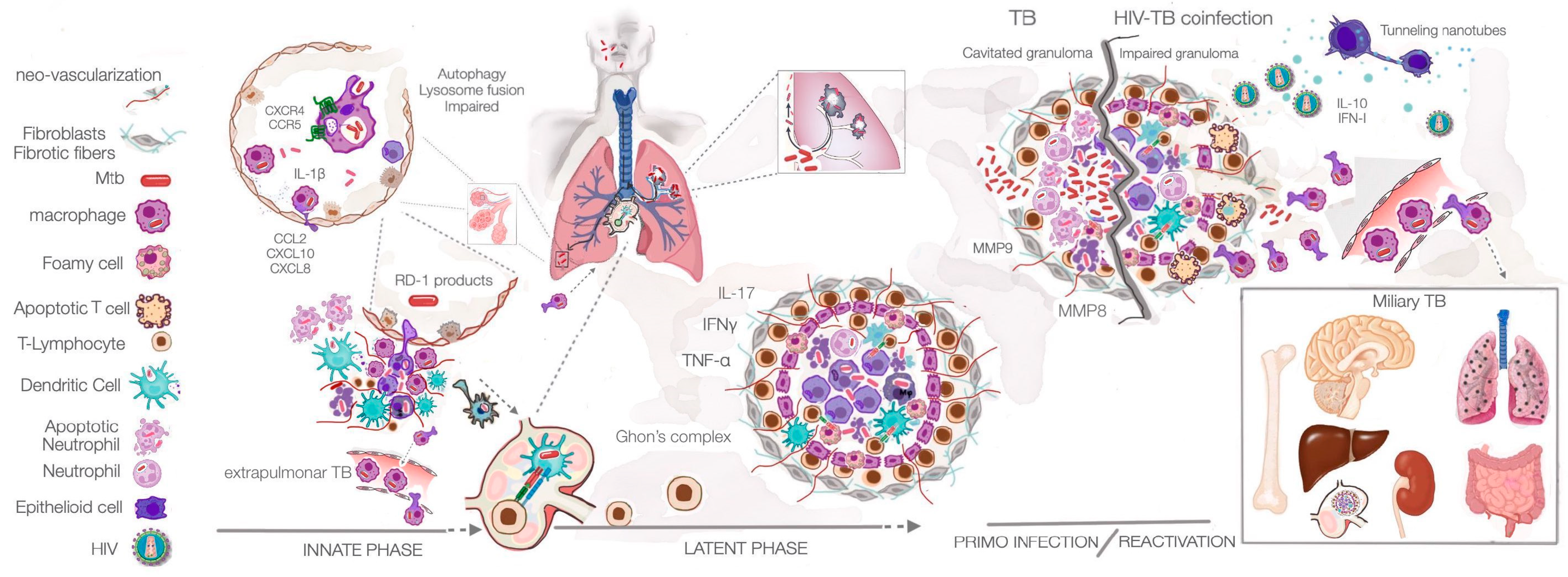
:1. Introduction
2. Main Consequences of HIV Infection
2.1. HIV as a Cytopathic Retrovirus
2.2. Establishment of Latently Infected Cells
2.3. Induction of Chronic Inflammation
3. Pathogenesis of Mycobacterium tuberculosis Infection
4. Consequences of HIV-MTB Co-Infection in the Amplification of Pathogenesis
4.1. Mtb infection in a Person with Pre-Existing HIV Infection
4.2. HIV Infection in a Person with Latent Mtb Infection

{kind=link}
| Cell or Tissue | Changes Induced by HIV Infection | References |
|---|---|---|
| CD4+ T lymphocytes | Apoptosis, which induces the death of infected and bystander cells; pyroptosis | [26,27,35] |
| Alveolar macrophages | Impaired phagosomal activity | [111,112] |
| Lung epithelial cells | Reduced expression of E-cadherin, which promotes paracellular permeability and triggers pro-inflammatory signals | [115] |
| Lung tissue | Shift in the cytokine microenvironment towards chemokine-driven networks involving SDF-1α, MIP-1α, MIP-1β, CCL2, CXCL10, GRO-α, eotaxin, and CXCL8 | [117] |
| NK | Reduced production of IFNγ, IL-15, and granzyme B in response to Mtb infection | [121] |
| Macrophages | The viral protein gp120 induces the production of IL-4 and IL-13, which are responsible for polarisation towards an anti-inflammatory M2 phenotype | [122,123] |
| Neutrophils | Death of Th17 CD4+ T lymphocytes impairs the recruitment of neutrophils and the development of necrotic granulomas | [142] |
| Alveolar macrophages | Nef viral protein enhances macrophage mesenchymal migration, facilitating viral spread to multiple organs | [146] |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2022 Factsheet. Available online: https://www.who.int/publications/m/item/global-tuberculosis-report-2022-factsheet (accessed on 15 January 2023).
- Anes, E.; Azevedo-Pereira, J.M.; Pires, D. Cathepsins and Their Endogenous Inhibitors in Host Defense During Mycobacterium tuberculosis and HIV Infection. Front. Immunol. 2021, 12, 726984. [Google Scholar] [CrossRef]
- Ramakrishnan, L. Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol. 2012, 12, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Cambier, C.J.; Falkow, S.; Ramakrishnan, L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell 2014, 159, 1497–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambier, C.J.; Takaki, K.K.; Larson, R.P.; Hernandez, R.E.; Tobin, D.M.; Urdahl, K.B.; Cosma, C.L.; Ramakrishnan, L. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 2014, 505, 218–222. [Google Scholar] [CrossRef] [Green Version]
- McKinney, J.D.; zu Bentrup, K.H.; Muñoz-Elías, E.J.; Miczak, A.; Chen, B.; Chan, W.-T.; Swenson, D.; Sacchettini, J.C.; Jacobs, W.R.; Russell, D.G. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 2000, 406, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Foreman, T.W.; Nelson, C.E.; Kauffman, K.D.; Lora, N.E.; Vinhaes, C.L.; Dorosky, D.E.; Sakai, S.; Gomez, F.; Fleegle, J.D.; Parham, M.; et al. CD4 T cells are rapidly depleted from tuberculosis granulomas following acute SIV co-infection. Cell Rep. 2022, 39, 110896. [Google Scholar] [CrossRef]
- WHO. HIV. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 25 January 2023).
- Brenchley, J.M.; Schacker, T.W.; Ruff, L.E.; Price, D.A.; Taylor, J.H.; Beilman, G.J.; Nguyen, P.L.; Khoruts, A.; Larson, M.; Haase, A.T.; et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 2004, 200, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Dufour, C.; Gantner, P.; Fromentin, R.; Chomont, N. The multifaceted nature of HIV latency. J. Clin. Investig. 2020, 130, 3381–3390. [Google Scholar] [CrossRef]
- Stevenson, M. HIV-1 pathogenesis. Nat. Med. 2003, 9, 853–860. [Google Scholar] [CrossRef]
- Zicari, S.; Sessa, L.; Cotugno, N.; Ruggiero, A.; Morrocchi, E.; Concato, C.; Rocca, S.; Zangari, P.; Manno, E.C.; Palma, P. Immune Activation, Inflammation, and Non-AIDS Co-Morbidities in HIV-Infected Patients under Long-Term ART. Viruses 2019, 11, 200. [Google Scholar] [CrossRef] [Green Version]
- Getahun, H.; Gunneberg, C.; Granich, R.; Nunn, P. HIV infection-associated tuberculosis: The epidemiology and the response. Clin. Infect. Dis. 2010, 50 (Suppl. S3), S201–S207. [Google Scholar] [CrossRef]
- Montales, M.T.; Chaudhury, A.; Beebe, A.; Patil, S.; Patil, N. HIV-Associated TB Syndemic: A Growing Clinical Challenge Worldwide. Front. Public Health 2015, 3, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, A.T. Targeting early infection to prevent HIV-1 mucosal transmission. Nature 2010, 464, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, A.G.; Beverley, P.C.; Clapham, P.R.; Crawford, D.H.; Greaves, M.F.; Weiss, R.A. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 1984, 312, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Klatzmann, D.R.; McDougal, J.S.; Maddon, P.J. The CD4 molecule and HIV infection. Immunodefic. Rev. 1990, 2, 43–66. [Google Scholar] [PubMed]
- Calado, M.; Matoso, P.; Santos-Costa, Q.; Espirito-Santo, M.; Machado, J.; Rosado, L.; Antunes, F.; Mansinho, K.; Lopes, M.M.; Maltez, F.; et al. Coreceptor usage by HIV-1 and HIV-2 primary isolates: The relevance of CCR8 chemokine receptor as an alternative coreceptor. Virology 2010, 408, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Simmons, G.; Reeves, J.D.; Hibbitts, S.; Stine, J.T.; Gray, P.W.; Proudfoot, A.E.; Clapham, P.R. Co-receptor use by HIV and inhibition of HIV infection by chemokine receptor ligands. Immunol. Rev. 2000, 177, 112–126. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Siliciano, R.F. In Vivo Dynamics of the Latent Reservoir for HIV-1: New Insights and Implications for Cure. Annu. Rev. Pathol. 2022, 17, 271–294. [Google Scholar] [CrossRef]
- Paiardini, M.; Müller-Trutwin, M. HIV-associated chronic immune activation. Immunol. Rev. 2013, 254, 78–101. [Google Scholar] [CrossRef] [Green Version]
- Cassol, E.; Alfano, M.; Biswas, P.; Poli, G. Monocyte-derived macrophages and myeloid cell lines as targets of HIV-1 replication and persistence. J. Leukoc. Biol. 2006, 80, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- McCune, J.M. The dynamics of CD4+ T-cell depletion in HIV disease. Nature 2001, 410, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.L.; Taylor, J.M.G.; Detels, R.; Hofmann, B.; Melmed, R.; Nishanian, P.; Giorgi, J.V. The Prognostic Value of Cellular and Serologic Markers in Infection with Human Immunodeficiency Virus Type 1. N. Engl. J. Med. 1990, 322, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Guadalupe, M.; Reay, E.; Sankaran, S.; Prindiville, T.; Flamm, J.; McNeil, A.; Dandekar, S. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J. Virol. 2003, 77, 11708–11717. [Google Scholar] [CrossRef] [Green Version]
- Laurent-Crawford, A.G.; Krust, B.; Muller, S.; Rivière, Y.; Rey-Cuille, M.A.; Béchet, J.M.; Montagnier, L.; Hovanessian, A.G. The cytopathic effect of HIV is associated with apoptosis. Virology 1991, 185, 829–839. [Google Scholar] [CrossRef]
- Finkel, T.H.; Tudor-Williams, G.; Banda, N.K.; Cotton, M.F.; Curiel, T.; Monks, C.; Baba, T.W.; Ruprecht, R.M.; Kupfer, A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1995, 1, 129–134. [Google Scholar] [CrossRef]
- Katsikis, P.D.; Wunderlich, E.S.; Smith, C.A.; Herzenberg, L.A.; Herzenberg, L.A. Fas antigen stimulation induces marked apoptosis of T lymphocytes in human immunodeficiency virus-infected individuals. J. Exp. Med. 1995, 181, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Herbeuval, J.-P.; Grivel, J.-C.; Boasso, A.; Hardy, A.W.; Chougnet, C.; Dolan, M.J.; Yagita, H.; Lifson, J.D.; Shearer, G.M. CD4+ T-cell death induced by infectious and noninfectious HIV-1: Role of type 1 interferon–dependent, TRAIL/DR5-mediated apoptosis. Blood 2005, 106, 3524–3531. [Google Scholar] [CrossRef] [PubMed]
- Schindler, M.; Munch, J.; Kutsch, O.; Li, H.; Santiago, M.L.; Bibollet-Ruche, F.; Müller-Trutwin, M.C.; Novembre, F.J.; Peeters, M.; Courgnaud, V.; et al. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell 2006, 125, 1055–1067. [Google Scholar] [CrossRef] [Green Version]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotič, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitaš, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Ali, A.; Arora, S.; Banerjea, A.C. Inhibition of {beta}-TrcP-dependent ubiquitination of p53 by HIV-1 Vpu promotes p53-mediated apoptosis in human T cells. Blood 2011, 117, 6600–6607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.L.; Le Rouzic, E.; Planelles, V. HIV-1 Vpr: Mechanisms of G2 arrest and apoptosis. Exp. Mol. Pathol. 2008, 85, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajasin, D.; Eugenin, E.A. HIV-1 Tat: Role in Bystander Toxicity. Front. Cell. Infect. Microbiol. 2020, 10, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doitsh, G.; Galloway, N.L.K.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Orenstein, J.M. In Vivo Cytolysis and Fusion of Human Immunodeficiency Virus Type 1-Infected Lymphocytes in Lymphoid Tissue. J. Infect. Dis. 2000, 182, 338–342. [Google Scholar] [CrossRef] [Green Version]
- Murooka, T.T.; Sharaf, R.R.; Mempel, T.R. Large Syncytia in Lymph Nodes Induced by CCR5-Tropic HIV-1. AIDS Res. Hum. Retrovir. 2015, 31, 471–472. [Google Scholar] [CrossRef] [Green Version]
- Symeonides, M.; Murooka, T.T.; Bellfy, L.N.; Roy, N.H.; Mempel, T.R.; Thali, M. HIV-1-Induced Small T Cell Syncytia Can Transfer Virus Particles to Target Cells through Transient Contacts. Viruses 2015, 7, 6590–6603. [Google Scholar] [CrossRef] [Green Version]
- Zack, J.A.; Arrigo, S.J.; Weitsman, S.R.; Go, A.S.; Haislip, A.; Chen, I.S. HIV-1 entry into quiescent primary lymphocytes: Molecular analysis reveals a labile, latent viral structure. Cell 1990, 61, 213–222. [Google Scholar] [CrossRef]
- Bukrinsky, M.I.; Stanwick, T.L.; Dempsey, M.P.; Stevenson, M. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 1991, 254, 423–427. [Google Scholar] [CrossRef]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Zhou, Y.; Kieffer, T.L.; Ruff, C.T.; Buck, C.; Siliciano, R.F. Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J. Virol. 2002, 76, 8518–8531. [Google Scholar] [CrossRef] [Green Version]
- Blankson, J.N.; Finzi, D.; Pierson, T.C.; Sabundayo, B.P.; Chadwick, K.; Margolick, J.B.; Quinn, T.C.; Siliciano, R.F. Biphasic decay of latently infected CD4+ T cells in acute human immunodeficiency virus type 1 infection. J. Infect. Dis. 2000, 182, 1636–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromentin, R.; Bakeman, W.; Lawani, M.B.; Khoury, G.; Hartogensis, W.; DaFonseca, S.; Killian, M.; Epling, L.; Hoh, R.; Sinclair, E.; et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS Pathog. 2016, 12, e1005761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGary, C.S.; Deleage, C.; Harper, J.; Micci, L.; Ribeiro, S.P.; Paganini, S.; Kuri-Cervantes, L.; Benne, C.; Ryan, E.S.; Balderas, R.; et al. CTLA-4(+)PD-1(-) Memory CD4(+) T Cells Critically Contribute to Viral Persistence in Antiretroviral Therapy-Suppressed, SIV-Infected Rhesus Macaques. Immunity 2017, 47, 776–788.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, V.A.; van der Sluis, R.M.; Solomon, A.; Dantanarayana, A.; McNeil, C.; Garsia, R.; Palmer, S.; Fromentin, R.; Chomont, N.; Sékaly, R.P.; et al. Programmed cell death-1 contributes to the establishment and maintenance of HIV-1 latency. AIDS 2018, 32, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Fromentin, R.; DaFonseca, S.; Costiniuk, C.T.; El-Far, M.; Procopio, F.A.; Hecht, F.M.; Hoh, R.; Deeks, S.G.; Hazuda, D.J.; Lewin, S.R.; et al. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4+ T cells from ART-suppressed individuals. Nat. Commun. 2019, 10, 814. [Google Scholar] [CrossRef] [Green Version]
- Uldrick, T.S.; Adams, S.V.; Fromentin, R.; Roche, M.; Fling, S.P.; Gonçalves, P.H.; Lurain, K.; Ramaswami, R.; Wang, C.-c.J.; Gorelick, R.J.; et al. Pembrolizumab induces HIV latency reversal in people living with HIV and cancer on antiretroviral therapy. Sci. Transl. Med. 2022, 14, eabl3836. [Google Scholar] [CrossRef]
- Gonzalez, S.M.; Aguilar-Jimenez, W.; Su, R.-C.; Rugeles, M.T. Mucosa: Key Interactions Determining Sexual Transmission of the HIV Infection. Front. Immunol. 2019, 10, 144. [Google Scholar] [CrossRef] [Green Version]
- Collman, R.G.; Yi, Y.; Liu, Q.H.; Freedman, B.D. Chemokine signaling and HIV-1 fusion mediated by macrophage CXCR4: Implications for target cell tropism. J. Leukoc. Biol. 2000, 68, 318–323. [Google Scholar] [CrossRef]
- Gorry, P.R.; Bristol, G.; Zack, J.A.; Ritola, K.; Swanstrom, R.; Birch, C.J.; Bell, J.E.; Bannert, N.; Crawford, K.; Wang, H.; et al. Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity. J. Virol. 2001, 75, 10073–10089. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.E.; Jaworowski, A.; Hearps, A.C. The HIV Reservoir in Monocytes and Macrophages. Front. Immunol. 2019, 10, 1435. [Google Scholar] [CrossRef] [Green Version]
- Kruize, Z.; Kootstra, N.A. The Role of Macrophages in HIV-1 Persistence and Pathogenesis. Front. Microbiol. 2019, 10, 2828. [Google Scholar] [CrossRef] [Green Version]
- Mukhamedova, N.; Hoang, A.; Dragoljevic, D.; Dubrovsky, L.; Pushkarsky, T.; Low, H.; Ditiatkovski, M.; Fu, Y.; Ohkawa, R.; Meikle, P.J.; et al. Exosomes containing HIV protein Nef reorganize lipid rafts potentiating inflammatory response in bystander cells. PLoS Pathogens 2019, 15, e1007907. [Google Scholar] [CrossRef] [Green Version]
- Roesch, F.; Richard, L.; Rua, R.; Porrot, F.; Casartelli, N.; Schwartz, O. Vpr Enhances Tumor Necrosis Factor Production by HIV-1-Infected T Cells. J. Virol. 2015, 89, 12118–12130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, E.P. The Role of Toll-Like Receptors in Retroviral Infection. Microorganisms 2020, 8, 1787. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Greene, W.C. Dissecting How CD4 T Cells Are Lost During HIV Infection. Cell Host Microbe 2016, 19, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Wacleche, V.S.; Landay, A.; Routy, J.P.; Ancuta, P. The Th17 Lineage: From Barrier Surfaces Homeostasis to Autoimmunity, Cancer, and HIV-1 Pathogenesis. Viruses 2017, 9, 303. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, A.M.; Brenchley, J.M. Microbial translocation: Translating simian immunodeficiency virus to HIV. Curr. Opin. HIV AIDS 2018, 13, 15–21. [Google Scholar] [CrossRef]
- Crakes, K.R.; Jiang, G. Gut Microbiome Alterations During HIV/SIV Infection: Implications for HIV Cure. Front. Microbiol. 2019, 10, 1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosadini, C.V.; Kagan, J.C. Early innate immune responses to bacterial LPS. Curr. Opin. Immunol. 2017, 44, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Babu, H.; Ambikan, A.T.; Gabriel, E.E.; Svensson Akusjärvi, S.; Palaniappan, A.N.; Sundaraj, V.; Mupanni, N.R.; Sperk, M.; Cheedarla, N.; Sridhar, R.; et al. Systemic Inflammation and the Increased Risk of Inflamm-Aging and Age-Associated Diseases in People Living With HIV on Long Term Suppressive Antiretroviral Therapy. Front. Immunol. 2019, 10, 1965. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.B.; Gern, B.H.; Delahaye, J.L.; Adams, K.N.; Plumlee, C.R.; Winkler, J.K.; Sherman, D.R.; Gerner, M.Y.; Urdahl, K.B. Alveolar Macrophages Provide an Early Mycobacterium tuberculosis Niche and Initiate Dissemination. Cell Host Microbe 2018, 24, 439–446.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.G.; VanderVen, B.C.; Glennie, S.; Mwandumba, H.; Heyderman, R.S. The macrophage marches on its phagosome: Dynamic assays of phagosome function. Nat. Rev. Immunol. 2009, 9, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.; Marques, J.; Pombo, J.P.; Carmo, N.; Bettencourt, P.; Neyrolles, O.; Lugo-Villarino, G.; Anes, E. Role of Cathepsins in Mycobacterium tuberculosis Survival in Human Macrophages. Sci. Rep. 2016, 6, 32247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.; Bernard, E.M.; Pombo, J.P.; Carmo, N.; Fialho, C.; Gutierrez, M.G.; Bettencourt, P.; Anes, E. Mycobacterium tuberculosis Modulates miR-106b-5p to Control Cathepsin S Expression Resulting in Higher Pathogen Survival and Poor T-Cell Activation. Front. Immunol. 2017, 8, 1819. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.; Calado, M.; Velez, T.; Mandal, M.; Catalão, M.J.; Neyrolles, O.; Lugo-Villarino, G.; Vérollet, C.; Azevedo-Pereira, J.M.; Anes, E. Modulation of Cystatin C in Human Macrophages Improves Anti-Mycobacterial Immune Responses to Mycobacterium tuberculosis Infection and Coinfection With HIV. Front. Immunol. 2021, 12, 4693. [Google Scholar] [CrossRef]
- Pires, D.; Valente, S.; Calado, M.; Mandal, M.; Azevedo-Pereira, J.M.; Anes, E. Repurposing Saquinavir for Host-Directed Therapy to Control Mycobacterium tuberculosis Infection. Front. Immunol. 2021, 12, 11. [Google Scholar] [CrossRef]
- Mwandumba, H.C.; Russell, D.G.; Nyirenda, M.H.; Anderson, J.; White, S.A.; Molyneux, M.E.; Squire, S.B. Mycobacterium tuberculosis Resides in Nonacidified Vacuoles in Endocytically Competent Alveolar Macrophages from Patients with Tuberculosis and HIV Infection1. J. Immunol. 2004, 172, 4592–4598. [Google Scholar] [CrossRef] [Green Version]
- Danilchanka, O.; Pires, D.; Anes, E.; Niederweis, M. The Mycobacterium tuberculosis Outer Membrane Channel Protein CpnT Confers Susceptibility to Toxic Molecules. Antimicrob. Agents Chemother. 2015, 59, 2328–2336. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.B.; Moura-Alves, P.; Sonawane, A.; Hacohen, N.; Griffiths, G.; Moita, L.F.; Anes, E. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell. Microbiol. 2010, 12, 1046–1063. [Google Scholar] [CrossRef]
- Anes, E.; Pires, D.; Mandal, M.; Azevedo-Pereira, J.M. Spatial localization of cathepsins: Implications in immune activation and resolution during infections. Front. Immunol. 2022, 13, 955407. [Google Scholar] [CrossRef]
- Russell, D.G. Who puts the tubercle in tuberculosis? Nat. Rev. Microbiol. 2007, 5, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, S.; Schaible, U. The Granuloma in Tuberculosis: Dynamics of a Host–Pathogen Collusion. Front. Immunol. 2013, 3, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagán, A.J.; Ramakrishnan, L. The Formation and Function of Granulomas. Annu. Rev. Immunol. 2018, 36, 639–665. [Google Scholar] [CrossRef]
- Peters, W.; Scott, H.M.; Chambers, H.F.; Flynn, J.L.; Charo, I.F.; Ernst, J.D. Chemokine receptor 2 serves an early and essential role in resistance to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2001, 98, 7958–7963. [Google Scholar] [CrossRef] [Green Version]
- Sadek, M.I.; Sada, E.; Toossi, Z.; Schwander, S.K.; Rich, E.A. Chemokines Induced by Infection of Mononuclear Phagocytes with Mycobacteria and Present in Lung Alveoli during Active Pulmonary Tuberculosis. Am. J. Respir. Cell Mol. Biol. 1998, 19, 513–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstein, M.; Schreiber, H.A.; Leiner, I.M.; Sušac, B.; Glickman, M.S.; Pamer, E.G. Essential yet limited role for CCR2+ inflammatory monocytes during Mycobacterium tuberculosis-specific T cell priming. eLife 2013, 2, e01086. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.J.; Linas, B.; Trevejo-Nuñez, G.J.; Kincaid, E.; Tamura, T.; Takatsu, K.; Ernst, J.D. Mycobacterium tuberculosis Infects Dendritic Cells with High Frequency and Impairs Their Function In Vivo. J. Immunol. 2007, 179, 2509–2519. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.J.; Desvignes, L.; Linas, B.; Banaiee, N.; Tamura, T.; Takatsu, K.; Ernst, J.D. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J. Exp. Med. 2007, 205, 105–115. [Google Scholar] [CrossRef]
- Harding, C.V.; Boom, W.H. Regulation of antigen presentation by Mycobacterium tuberculosis: A role for Toll-like receptors. Nat. Rev. Microbiol. 2010, 8, 296–307. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Ernst, J.D.; Desvignes, L. Beyond macrophages: The diversity of mononuclear cells in tuberculosis. Immunol. Rev. 2014, 262, 179–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, T.A.; Koch, M.; Nouailles, G.; Jacobsen, M.; Kosmiadi, G.A.; Miekley, D.; Kuhlmann, S.; Jörg, S.; Gamradt, P.; Mollenkopf, H.-J.; et al. Secondary lymphoid organs are dispensable for the development of T-cell-mediated immunity during tuberculosis. Eur. J. Immunol. 2010, 40, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.M. T cells in mycobacterial infection and disease. Curr. Opin. Immunol. 2009, 21, 378–384. [Google Scholar] [CrossRef] [Green Version]
- Ulrichs, T.; Kaufmann, S.H.E. New insights into the function of granulomas in human tuberculosis. J. Pathol. 2006, 208, 261–269. [Google Scholar] [CrossRef]
- Balasubramanian, V.; Wiegeshaus, E.H.; Taylor, B.T.; Smith, D.W. Pathogenesis of tuberculosis: Pathway to apical localization. Tuber. Lung Dis. 1994, 75, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, M.; Kaufmann, S.H.E. Mycobacterium tuberculosis: Success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–532. [Google Scholar] [CrossRef] [Green Version]
- Ehlers, S. Role of tumour necrosis factor (TNF) in host defence against tuberculosis: Implications for immunotherapies targeting TNF. Ann. Rheum. Dis. 2003, 62, ii37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacMicking, J.D. Cell-Autonomous Effector Mechanisms against Mycobacterium tuberculosis. Cold Spring Harb. Perspect. Med. 2014, 4, a018507. [Google Scholar] [CrossRef] [Green Version]
- Schnettger, L.; Rodgers, A.; Repnik, U.; Lai, R.P.; Pei, G.; Verdoes, M.; Wilkinson, R.J.; Young, D.B.; Gutierrez, M.G. A Rab20-Dependent Membrane Trafficking Pathway Controls M. tuberculosis Replication by Regulating Phagosome Spaciousness and Integrity. Cell Host Microbe 2017, 21, 619–628.e5. [Google Scholar] [CrossRef]
- Roach, D.R.; Bean, A.G.D.; Demangel, C.; France, M.P.; Briscoe, H.; Britton, W.J. TNF Regulates Chemokine Induction Essential for Cell Recruitment, Granuloma Formation, and Clearance of Mycobacterial Infection1. J. Immunol. 2002, 168, 4620–4627. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.M.; Dalton, D.K.; Stewart, T.A.; Griffin, J.P.; Russell, D.G.; Orme, I.M. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 1993, 178, 2243–2247. [Google Scholar] [CrossRef] [Green Version]
- Blanchette, J.; Jaramillo, M.; Olivier, M. Signalling events involved in interferon-γ-inducible macrophage nitric oxide generation. Immunology 2003, 108, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.A.; Feng, C.G.; Sher, A. p47 GTPases: Regulators of immunity to intracellular pathogens. Nat. Rev. Immunol. 2004, 4, 100–109. [Google Scholar] [CrossRef]
- Desvignes, L.; Ernst, J.D. Interferon-γ-Responsive Nonhematopoietic Cells Regulate the Immune Response to Mycobacterium tuberculosis. Immunity 2009, 31, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, G.; Post Frank, A.; Moreira Andre, L.; Wainwright, H.; Kreiswirth Barry, N.; Tanverdi, M.; Mathema, B.; Ramaswamy Srinivas, V.; Walther, G.; Steyn Lafras, M.; et al. Mycobacterium tuberculosis Growth at theCavity Surface: A Microenvironment with FailedImmunity. Infect. Immun. 2003, 71, 7099–7108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dheda, K.; Booth, H.; Huggett, J.F.; Johnson, M.A.; Zumla, A.; Rook, G.A.W. Lung Remodeling in Pulmonary Tuberculosis. J. Infect. Dis. 2005, 192, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Reece, S.T.; Kaufmann, S.H.E. Floating between the poles of pathology and protection: Can we pin down the granuloma in tuberculosis? Curr. Opin. Microbiol. 2012, 15, 63–70. [Google Scholar] [CrossRef]
- CDC. Pneumocystis pneumonia--Los Angeles. MMWR Morb. Mortal. Wkly. Rep. 1981, 30, 250–252. [Google Scholar]
- Feldman, C.; Anderson, R. HIV-Associated Bacterial Pneumonia. Clin. Chest Med. 2013, 34, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, R.; Uldrick, T.S. HIV-Associated Cancers and Related Diseases. N. Engl. J. Med. 2018, 378, 1029–1041. [Google Scholar] [CrossRef]
- Jambo, K.C.; Banda, D.H.; Kankwatira, A.M.; Sukumar, N.; Allain, T.J.; Heyderman, R.S.; Russell, D.G.; Mwandumba, H.C. Small alveolar macrophages are infected preferentially by HIV and exhibit impaired phagocytic function. Mucosal Immunol. 2014, 7, 1116–1126. [Google Scholar] [CrossRef]
- Cribbs, S.K.; Caliendo, A.M.; Guidot, D.M. Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res. Hum. Retrovir. 2015, 31, 64–70. [Google Scholar] [CrossRef]
- Schiff, A.E.; Linder, A.H.; Luhembo, S.N.; Banning, S.; Deymier, M.J.; Diefenbach, T.J.; Dickey, A.K.; Tsibris, A.M.; Balazs, A.B.; Cho, J.L.; et al. T cell-tropic HIV efficiently infects alveolar macrophages through contact with infected CD4+ T cells. Sci. Rep. 2021, 11, 3890. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Meshesha, M.; Churgulia, N.; Merlo, C.; Fuchs, E.; Breakey, J.; Jones, J.; Stivers, J.T. Replication-competent HIV-1 in human alveolar macrophages and monocytes despite nucleotide pools with elevated dUTP. Retrovirology 2022, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- Alimohammadi, A.; Coker, R.; Miller, R.; Mitchell, D.; Williamson, J.; Clarke, J. Genotypic variants of HIV-1 from peripheral blood and lungs of AIDS patients. AIDS 1997, 11, 831–832. [Google Scholar] [PubMed]
- Silviu, I.; Paul, F.S.; Robert, J.W.; Harold, S.G. Human Immunodeficiency Virus Type 1 Strains in the Lungs of Infected Individuals Evolve Independently from Those in Peripheral Blood and are Highly Conserved in the C-Terminal Region of the Envelope V3 Loop. Proc. Natl. Acad. Sci. USA 1994, 91, 11378–11382. [Google Scholar] [CrossRef] [Green Version]
- van’t Wout, A.B.; Ran, L.J.; Kuiken, C.L.; Kootstra, N.A.; Pals, S.T.; Schuitemaker, H. Analysis of the temporal relationship between human immunodeficiency virus type 1 quasispecies in sequential blood samples and various organs obtained at autopsy. J. Virol. 1998, 72, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzolini, J.; Herit, F.; Bouchet, J.; Benmerah, A.; Benichou, S.; Niedergang, F. Inhibition of phagocytosis in HIV-1-infected macrophages relies on Nef-dependent alteration of focal delivery of recycling compartments. Blood 2010, 115, 4226–4236. [Google Scholar] [CrossRef] [Green Version]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Kedzierska, K.; Azzam, R.; Ellery, P.; Mak, J.; Jaworowski, A.; Crowe, S.M. Defective phagocytosis by human monocyte/macrophages following HIV-1 infection: Underlying mechanisms and modulation by adjunctive cytokine therapy. J. Clin. Virol. 2003, 26, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Devadoss, D.; Singh, S.P.; Acharya, A.; Do, K.C.; Periyasamy, P.; Manevski, M.; Mishra, N.; Tellez, C.S.; Ramakrishnan, S.; Belinsky, S.A.; et al. HIV-1 Productively Infects and Integrates in Bronchial Epithelial Cells. Front. Cell. Infect. Microbiol. 2020, 10, 612360. [Google Scholar] [CrossRef] [PubMed]
- Brune, K.A.; Ferreira, F.; Mandke, P.; Chau, E.; Aggarwal, N.R.; D’Alessio, F.R.; Lambert, A.A.; Kirk, G.; Blankson, J.; Drummond, M.B.; et al. HIV Impairs Lung Epithelial Integrity and Enters the Epithelium to Promote Chronic Lung Inflammation. PLoS ONE 2016, 11, e0149679. [Google Scholar] [CrossRef]
- Head, B.M.; Mao, R.; Keynan, Y.; Rueda, Z.V. Inflammatory mediators and lung abnormalities in HIV: A systematic review. PLoS ONE 2019, 14, e0226347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jambo, K.C.; Tembo, D.L.; Kamng’ona, A.W.; Musicha, P.; Banda, D.H.; Kankwatira, A.M.; Malamba, R.D.; Allain, T.J.; Heyderman, R.S.; Russell, D.G.; et al. HIV-associated disruption of lung cytokine networks is incompletely restored in asymptomatic HIV-infected Malawian adults on antiretroviral therapy. ERJ Open Res. 2017, 3, 00097–02017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucşan, A.N.; Chatterjee, A.; Singh, D.K.; Foreman, T.W.; Lee, T.-H.; Threeton, B.; Kirkpatrick, M.G.; Ahmed, M.; Golden, N.; Alvarez, X.; et al. Mechanisms of reactivation of latent tuberculosis infection due to SIV coinfection. J. Clin. Investig. 2019, 129, 5254–5260. [Google Scholar] [CrossRef] [PubMed]
- Esmail, H.; Riou, C.; du Bruyn, E.; Lai, R.P.-J.; Harley, Y.X.R.; Meintjes, G.; Wilkinson, K.A.; Wilkinson, R.J. The Immune Response to Mycobacterium tuberculosis in HIV-1-Coinfected Persons. Annu. Rev. Immunol. 2018, 36, 603–638. [Google Scholar] [CrossRef]
- Devalraju, K.P.; Neela, V.S.K.; Krovvidi, S.S.; Vankayalapati, R.; Valluri, V.L. Defective expansion and function of memory like natural killer cells in HIV+ individuals with latent tuberculosis infection. PLoS ONE 2021, 16, e0257185. [Google Scholar] [CrossRef]
- Hirsch, C.S.; Toossi, Z.; Othieno, C.; Johnson, J.L.; Schwander, S.K.; Robertson, S.; Wallis, R.S.; Edmonds, K.; Okwera, A.; Mugerwa, R.; et al. Depressed T-cell interferon-gamma responses in pulmonary tuberculosis: Analysis of underlying mechanisms and modulation with therapy. J. Infect. Dis. 1999, 180, 2069–2073. [Google Scholar] [CrossRef] [Green Version]
- Patella, V.; Florio, G.; Petraroli, A.; Marone, G. HIV-1 gp120 induces IL-4 and IL-13 release from human Fc epsilon RI+ cells through interaction with the VH3 region of IgE. J. Immunol. 2000, 164, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Olson, G.S.; Murray, T.A.; Jahn, A.N.; Mai, D.; Diercks, A.H.; Gold, E.S.; Aderem, A. Type I interferon decreases macrophage energy metabolism during mycobacterial infection. Cell Rep. 2021, 35, 109195. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Padmapriyadarsini, C.; Ponnuraja, C.; Sumathi, C.H.; Rajasekaran, S.; Amerandran, V.A.; Reddy, M.; Deivanayagam, C.N. Miliary tuberculosis in human immunodeficiency virus infected patients not on antiretroviral therapy: Clinical profile and response to shortcourse chemotherapy. J. Postgrad. Med. 2007, 53, 228–231. [Google Scholar] [CrossRef]
- Whalen, C.; Horsburgh, C.R.; Hom, D.; Lahart, C.; Simberkoff, M.; Ellner, J. Accelerated course of human immunodeficiency virus infection after tuberculosis. Am. J. Respir. Crit. Care Med. 1995, 151, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, Z.A.; Wong, E.B.; Ndung’u, T.; Kasprowicz, V.O.; Bishai, W.R. Latent and Active Tuberculosis Infection Increase Immune Activation in Individuals Co-Infected with HIV. eBioMedicine 2015, 2, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goletti, D.; Weissman, D.; Jackson, R.W.; Graham, N.M.; Vlahov, D.; Klein, R.S.; Munsiff, S.S.; Ortona, L.; Cauda, R.; Fauci, A.S. Effect of Mycobacterium tuberculosis on HIV replication. Role of immune activation. J. Immunol. 1996, 157, 1271–1278. [Google Scholar] [CrossRef]
- Nakata, K.; Rom, W.N.; Honda, Y.; Condos, R.; Kanegasaki, S.; Cao, Y.; Weiden, M. Mycobacterium tuberculosis enhances human immunodeficiency virus-1 replication in the lung. Am. J. Respir. Crit. Care Med. 1997, 155, 996–1003. [Google Scholar] [CrossRef]
- Lawn, S.D.; Pisell, T.L.; Hirsch, C.S.; Wu, M.; Butera, S.T.; Toossi, Z. Anatomically Compartmentalized Human Immunodeficiency Virus Replication in HLA-DR+ Cells and CD14+ Macrophages at the Site of Pleural Tuberculosis Coinfection. J. Infect. Dis. 2001, 184, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Matthews, K.; Ntsekhe, M.; Syed, F.; Scriba, T.; Russell, J.; Tibazarwa, K.; Deffur, A.; Hanekom, W.; Mayosi, B.M.; Wilkinson, R.J.; et al. HIV-1 infection alters CD4+ memory T-cell phenotype at the site of disease in extrapulmonary tuberculosis. Eur. J. Immunol. 2012, 42, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Rosas-Taraco, A.G.; Arce-Mendoza, A.Y.; Caballero-Olín, G.; Salinas-Carmona, M.C. Mycobacterium tuberculosis upregulates coreceptors CCR5 and CXCR4 while HIV modulates CD14 favoring concurrent infection. AIDS Res. Hum. Retrovir. 2006, 22, 45–51. [Google Scholar] [CrossRef]
- Bernier, R.; Barbeau, B.; Olivier, M.; Tremblay, M.J. Mycobacterium tuberculosis mannose-capped lipoarabinomannan can induce NF-kappaB-dependent activation of human immunodeficiency virus type 1 long terminal repeat in T cells. J. Gen. Virol. 1998, 79, 1353–1361. [Google Scholar] [CrossRef] [Green Version]
- Israël-Biet, D.; Cadranel, J.; Beldjord, K.; Andrieu, J.M.; Jeffrey, A.; Even, P. Tumor necrosis factor production in HIV-seropositive subjects. Relationship with lung opportunistic infections and HIV expression in alveolar macrophages. J. Immunol. 1991, 147, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, K.; Crowe, S.M.; Turville, S.; Cunningham, A.L. The influence of cytokines, chemokines and their receptors on HIV-1 replication in monocytes and macrophages. Rev. Med. Virol. 2003, 13, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, C.; Ngwenyama, N.; Schuetz, A.; Petrovas, C.; Reither, K.; Heeregrave, E.J.; Casazza, J.P.; Ambrozak, D.R.; Louder, M.; Ampofo, W.; et al. Preferential infection and depletion of Mycobacterium tuberculosis–specific CD4 T cells after HIV-1 infection. J. Exp. Med. 2010, 207, 2869–2881. [Google Scholar] [CrossRef] [Green Version]
- Corleis, B.; Bucsan, A.N.; Deruaz, M.; Vrbanac, V.D.; Lisanti-Park, A.C.; Gates, S.J.; Linder, A.H.; Paer, J.M.; Olson, G.S.; Bowman, B.A.; et al. HIV-1 and SIV Infection Are Associated with Early Loss of Lung Interstitial CD4+ T Cells and Dissemination of Pulmonary Tuberculosis. Cell Rep. 2019, 26, 1409–1418.e5. [Google Scholar] [CrossRef] [Green Version]
- Neff, C.P.; Atif, S.M.; Logue, E.C.; Siebert, J.; Görg, C.; Lavelle, J.; Fiorillo, S.; Twigg, H.; Campbell, T.B.; Fontenot, A.P.; et al. HIV Infection Is Associated with Loss of Anti-Inflammatory Alveolar Macrophages. J. Immunol. 2020, 205, 2447–2455. [Google Scholar] [CrossRef]
- Chung, N.P.Y.; Khan, K.M.F.; Kaner, R.J.; O’Beirne, S.L.; Crystal, R.G. HIV induces airway basal progenitor cells to adopt an inflammatory phenotype. Sci. Rep. 2021, 11, 3988. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Eddy, J.J.; Jacobson, K.R.; Henderson, A.J.; Agosto, L.M. Enhanced Human Immunodeficiency Virus-1 Replication in CD4+ T Cells Derived From Individuals With Latent Mycobacterium tuberculosis Infection. J. Infect. Dis. 2020, 222, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Tse, D.B.; Rochford, G.; Prabhakar, S.; Hoshino, S.; Chitkara, N.; Kuwabara, K.; Ching, E.; Raju, B.; Gold, J.A.; et al. Mycobacterium tuberculosis-Induced CXCR4 and Chemokine Expression Leads to Preferential X4 HIV-1 Replication in Human Macrophages1. J. Immunol. 2004, 172, 6251–6258. [Google Scholar] [CrossRef] [Green Version]
- Juffermans, N.P.; Speelman, P.; Verbon, A.; Veenstra, J.; Jie, C.; van Deventer, S.J.; van Der Poll, T. Patients with active tuberculosis have increased expression of HIV coreceptors CXCR4 and CCR5 on CD4(+) T cells. Clin. Infect. Dis. 2001, 32, 650–652. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.E. Future Vaccination Strategies against Tuberculosis: Thinking outside the Box. Immunity 2010, 33, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Dorhoi, A.; Kaufmann, S.H.E. Pathology and immune reactivity: Understanding multidimensionality in pulmonary tuberculosis. Semin. Immunopathol. 2016, 38, 153–166. [Google Scholar] [CrossRef]
- Huang, C.-C.; Tchetgen, E.T.; Becerra, M.C.; Cohen, T.; Hughes, K.C.; Zhang, Z.; Calderon, R.; Yataco, R.; Contreras, C.; Galea, J.; et al. The Effect of HIV-Related Immunosuppression on the Risk of Tuberculosis Transmission to Household Contacts. Clin. Infect. Dis. 2014, 58, 765–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaron, L.; Saadoun, D.; Calatroni, I.; Launay, O.; Mémain, N.; Vincent, V.; Marchal, G.; Dupont, B.; Bouchaud, O.; Valeyre, D.; et al. Tuberculosis in HIV-infected patients: A comprehensive review. Clin. Microbiol. Infect. 2004, 10, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Vérollet, C.; Souriant, S.; Bonnaud, E.; Jolicoeur, P.; Raynaud-Messina, B.; Kinnaer, C.; Fourquaux, I.; Imle, A.; Benichou, S.; Fackler, O.T.; et al. HIV-1 reprograms the migration of macrophages. Blood 2015, 125, 1611–1622. [Google Scholar] [CrossRef] [Green Version]
- Seddon, J.A.; Chiang, S.S.; Esmail, H.; Coussens, A.K. The Wonder Years: What Can Primary School Children Teach Us About Immunity to Mycobacterium tuberculosis? Front. Immunol. 2018, 9, 2946. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandler, N.G.; Douek, D.C. Microbial translocation in HIV infection: Causes, consequences and treatment opportunities. Nat. Rev. Microbiol. 2012, 10, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Shankar, E.M.; Vignesh, R.; Ellegård, R.; Barathan, M.; Chong, Y.K.; Bador, M.K.; Rukumani, D.V.; Sabet, N.S.; Kamarulzaman, A.; Velu, V.; et al. HIV-Mycobacterium tuberculosis co-infection: A “danger-couple model” of disease pathogenesis. Pathog. Dis. 2014, 70, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, A.; Villa, S.; Castelli, V.; Bandera, A.; Gori, A. T-Cell Exhaustion in Mycobacterium tuberculosis and Nontuberculous Mycobacteria Infection: Pathophysiology and Therapeutic Perspectives. Microorganisms 2021, 9, 2460. [Google Scholar] [CrossRef]
- Mayer-Barber, K.D.; Andrade, B.B.; Oland, S.D.; Amaral, E.P.; Barber, D.L.; Gonzales, J.; Derrick, S.C.; Shi, R.; Kumar, N.P.; Wei, W.; et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014, 511, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Mourik, B.C.; Lubberts, E.; de Steenwinkel, J.E.M.; Ottenhoff, T.H.M.; Leenen, P.J.M. Interactions between Type 1 Interferons and the Th17 Response in Tuberculosis: Lessons Learned from Autoimmune Diseases. Front. Immunol. 2017, 8, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassermann, R.; Gulen, M.F.; Sala, C.; Perin, S.G.; Lou, Y.; Rybniker, J.; Schmid-Burgk, J.L.; Schmidt, T.; Hornung, V.; Cole, S.T.; et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host & Microbe 2015, 17, 799–810. [Google Scholar] [CrossRef] [Green Version]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Naujoks, J.; Tabeling, C.; Dill, B.D.; Hoffmann, C.; Brown, A.S.; Kunze, M.; Kempa, S.; Peter, A.; Mollenkopf, H.-J.; Dorhoi, A.; et al. IFNs Modify the Proteome of Legionella-Containing Vacuoles and Restrict Infection Via IRG1-Derived Itaconic Acid. PLoS Pathogens 2016, 12, e1005408. [Google Scholar] [CrossRef] [Green Version]
- Murira, A.; Lamarre, A. Type-I Interferon Responses: From Friend to Foe in the Battle against Chronic Viral Infection. Front. Immunol. 2016, 7, 609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Kang, W.; Zuo, J.; Kang, W.; Sun, Y. The Significance of Type-I Interferons in the Pathogenesis and Therapy of Human Immunodeficiency Virus 1 Infection. Front. Immunol. 2017, 8, 1431. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Teixeira, L.; Mayer-Barber, K.; Sher, A.; O’Garra, A. Type I interferons in tuberculosis: Foe and occasionally friend. J. Exp. Med. 2018, 215, 1273–1285. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Teixeira, L.; Stimpson, P.J.; Stavropoulos, E.; Hadebe, S.; Chakravarty, P.; Ioannou, M.; Aramburu, I.V.; Herbert, E.; Priestnall, S.L.; Suarez-Bonnet, A.; et al. Type I IFN exacerbates disease in tuberculosis-susceptible mice by inducing neutrophil-mediated lung inflammation and NETosis. Nat. Commun. 2020, 11, 5566. [Google Scholar] [CrossRef]
- Donovan, M.L.; Schultz, T.E.; Duke, T.J.; Blumenthal, A. Type I Interferons in the Pathogenesis of Tuberculosis: Molecular Drivers and Immunological Consequences. Front. Immunol. 2017, 8, 1633. [Google Scholar] [CrossRef] [Green Version]
- Herbein, G.; Varin, A. The macrophage in HIV-1 infection: From activation to deactivation? Retrovirology 2010, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, M.; Souriant, S.; Balboa, L.; Vu Manh, T.-P.; Pingris, K.; Rousset, S.; Cougoule, C.; Rombouts, Y.; Poincloux, R.; Ben Neji, M.; et al. Tuberculosis-associated IFN-I induces Siglec-1 on tunneling nanotubes and favors HIV-1 spread in macrophages. eLife 2020, 9, e52535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souriant, S.; Balboa, L.; Dupont, M.; Pingris, K.; Kviatcovsky, D.; Cougoule, C.; Lastrucci, C.; Bah, A.; Gasser, R.; Poincloux, R.; et al. Tuberculosis Exacerbates HIV-1 Infection through IL-10/STAT3-Dependent Tunneling Nanotube Formation in Macrophages. Cell Rep. 2019, 26, 3586–3599.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, R.P.; Meintjes, G.; Wilkinson, R.J. HIV-1 tuberculosis-associated immune reconstitution inflammatory syndrome. Semin. Immunopathol. 2016, 38, 185–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| Cell or Tissue | Changes Induced by Mycobacterium tuberculosis (Mtb) Infection | References |
|---|---|---|
| Peripheral blood | Immune responses to Mtb contribute to increased HIV replication through the activation of macrophages and CD4+ T lymphocytes | [127] |
| Lungs | Immune responses to Mtb contribute to increased HIV replication through the activation of macrophages and CD4+ T lymphocytes | [128] |
| Granuloma | Recruitment of CD4+ T lymphocyte and macrophage populations expressing membrane receptors required for viral infection | [131,139,140,141] |
| Alveolar and interstitial macrophages | Induces the expression of HIV coreceptors CCR5 and CXCR4, promoting viral infection of these cells | [131] |
| Macrophages and CD4+ T lymphocytes | The Mtb wall glycolipid LAM induces the secretion of pro-inflammatory cytokines that activate transcription factors in CD4+ T lymphocytes and macrophages harbouring proviral DNA, leading to the transcriptional activation of integrated proviral DNA and the production of new viral particles | [131,132,133,134] |
| Macrophages and dendritic cells | Increased expression of IDO, leading to an anergic state in T lymphocytes, with the concomitant decreased secretion of IFNγ, which promotes viral replication | [148,149,150] |
| Several compartments | Prolonged secretion of IFN-I, which induces the polarisation of pro-inflammatory macrophages and dendritic cells into an immune-deactivated state via immunosuppressive cytokines such as IL-10 and TGFβ. Both cytokines are associated with fibrosis in lymph nodes, impairing their function and contributing to immunodeficiency and AIDS progression | [152,153,154,163] |
| Macrophages | Promotes direct cell-to-cell viral transfer through the formation of tunnelling nanotubes induced in macrophages stimulated by IL-10 and IFN-I | [164,165] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azevedo-Pereira, J.M.; Pires, D.; Calado, M.; Mandal, M.; Santos-Costa, Q.; Anes, E. HIV/Mtb Co-Infection: From the Amplification of Disease Pathogenesis to an “Emerging Syndemic”. Microorganisms 2023, 11, 853. https://doi.org/10.3390/microorganisms11040853
Azevedo-Pereira JM, Pires D, Calado M, Mandal M, Santos-Costa Q, Anes E. HIV/Mtb Co-Infection: From the Amplification of Disease Pathogenesis to an “Emerging Syndemic”. Microorganisms. 2023; 11(4):853. https://doi.org/10.3390/microorganisms11040853
Chicago/Turabian StyleAzevedo-Pereira, José Miguel, David Pires, Marta Calado, Manoj Mandal, Quirina Santos-Costa, and Elsa Anes. 2023. "HIV/Mtb Co-Infection: From the Amplification of Disease Pathogenesis to an “Emerging Syndemic”" Microorganisms 11, no. 4: 853. https://doi.org/10.3390/microorganisms11040853