

Characterization of the Bacterial Microbiome in Natural Populations of Barley Stem Gall Midge, Mayetiola hordei, in Morocco

 ,
,  , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Barley Stem Gall Midge Collection and DNA Isolation
2.2. Screening and Identification of Bacterial Symbionts
2.3. Amplification of the V3-V4 Region, Index PCR, Purification, and Illumina Sequencing
2.4. NGS Sequencing and Bioinformatics Analysis
2.5. Phylogenetic Analysis
2.6. Core Microbiome
3. Results
3.1. Infection Status of Reproductive Symbionts in Natural Populations of BSGM
3.1.1. Infection Prevalence
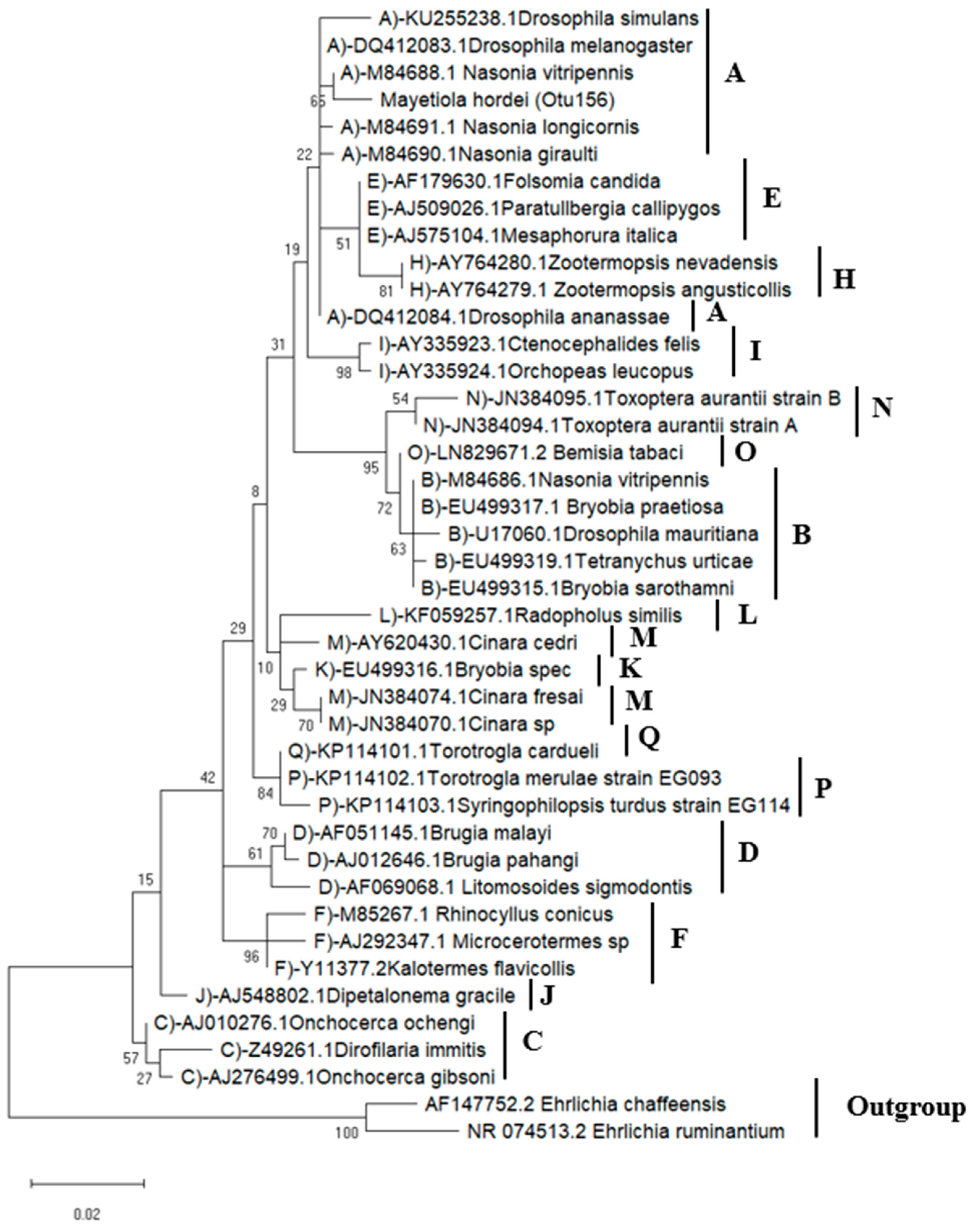
3.1.2. Phylogenetic Analysis of Wolbachia Sequences in BSGM Populations
3.2. 16S rRNA Amplicon Sequencing
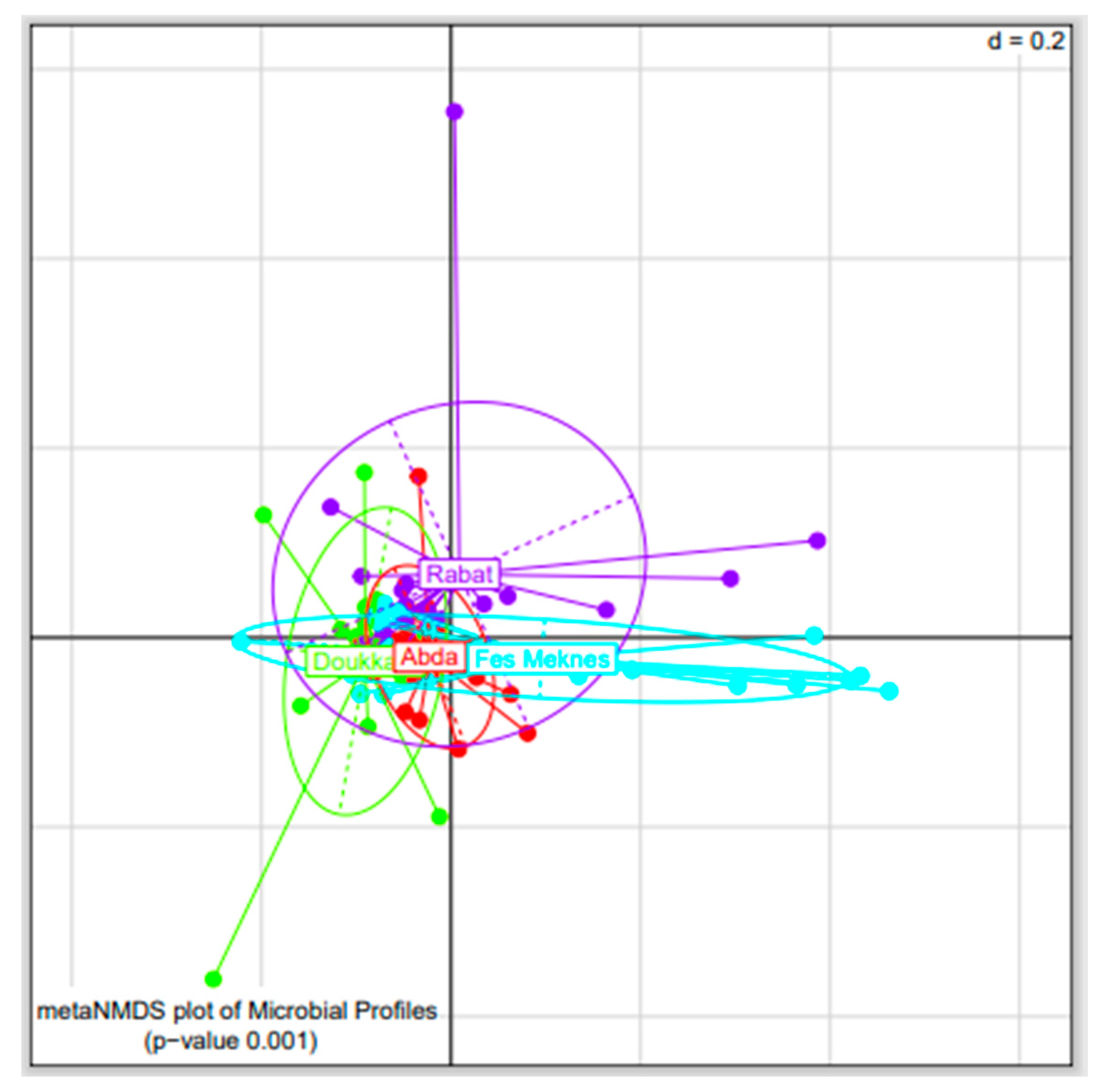
3.2.1. Bacterial Diversity among BSGM Natural Populations
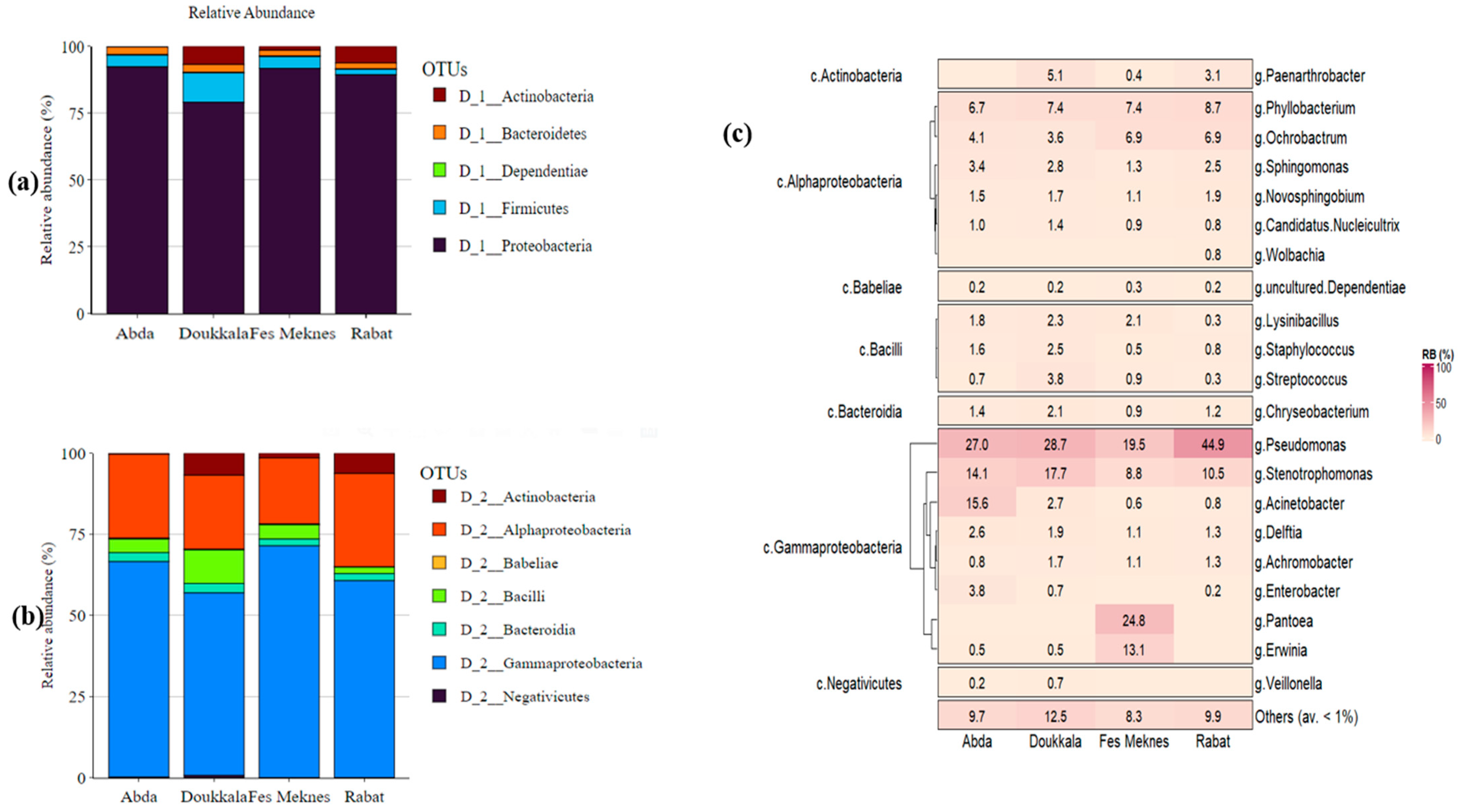
3.2.2. Bacterial Composition of BSGM Natural Populations
3.2.3. Wolbachia Phylogenetic Analysis
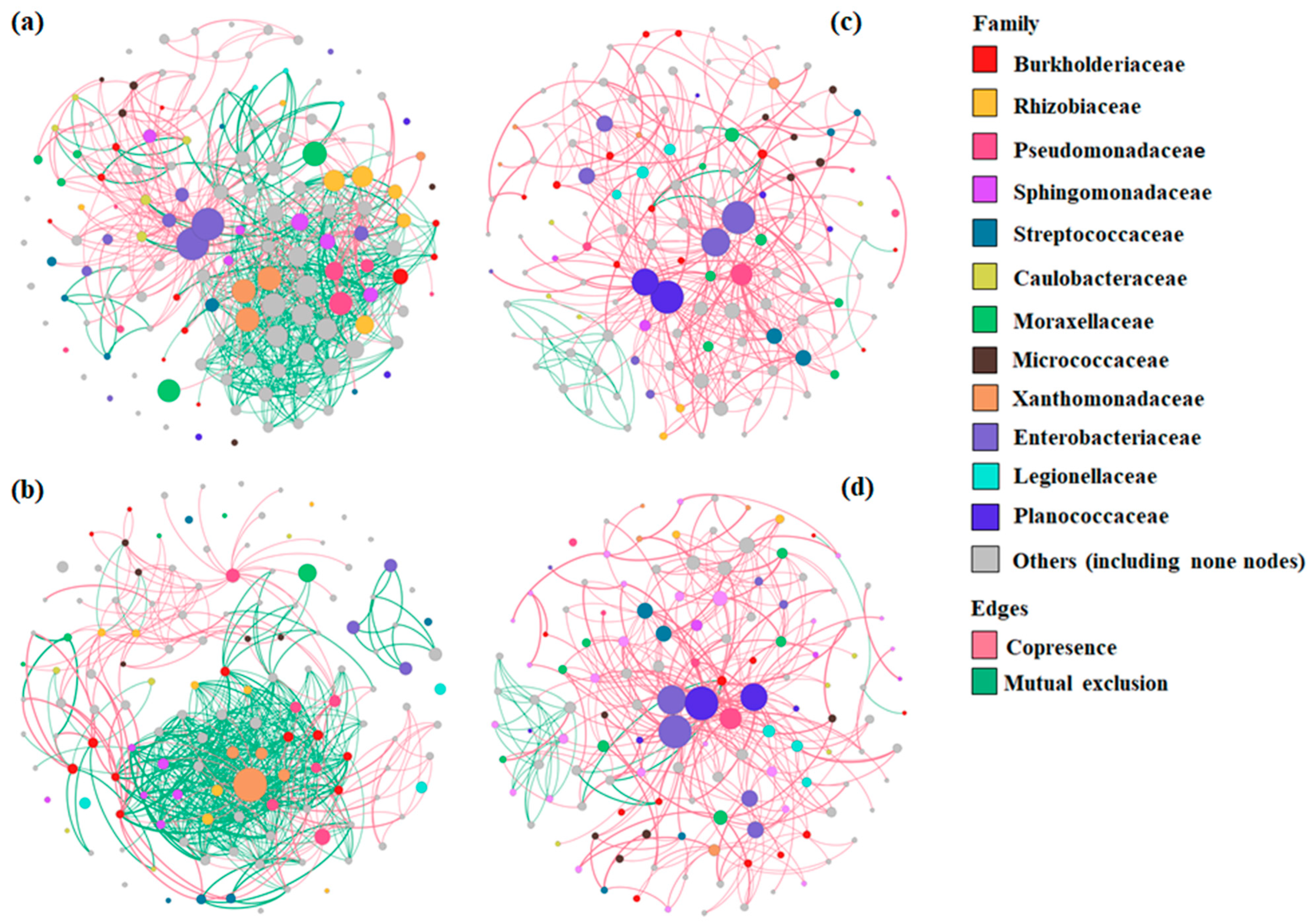
3.2.4. Bacterial Co-Occurrence/Mutual Exclusion Network Analysis of BSGM Natural Populations
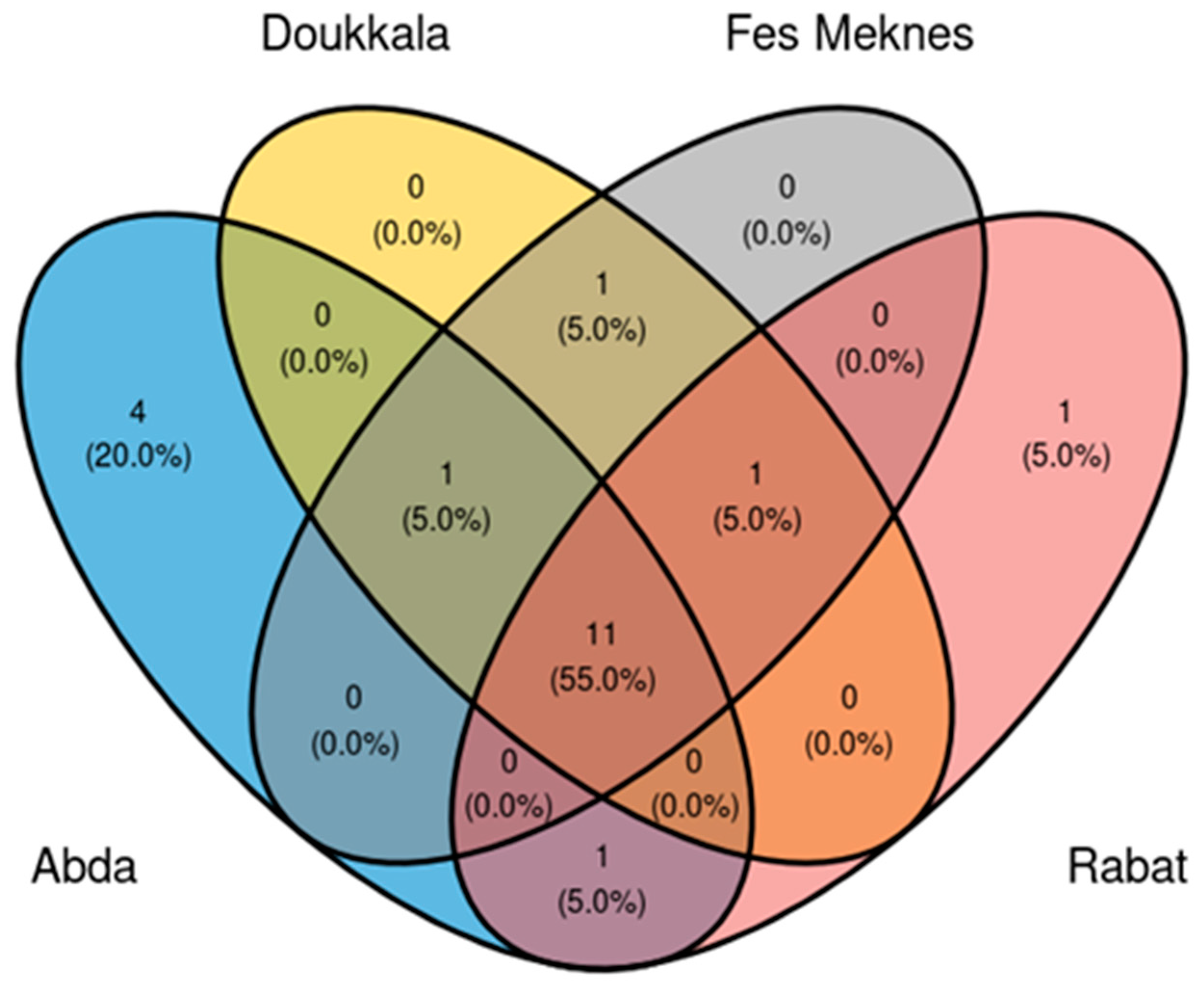
3.2.5. Core Microbiome Analysis of BSGM Populations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cherif, A.; Mediouni Ben Jemâa, J. Distribution, population dynamics and damage potential of barley stem gall midge, Mayetiola hordei (Diptera: Cecidomyiidae) on cultivated barley in two semi-arid areas of North Tunisia. Crop Prot. 2018, 112, 295–303. [Google Scholar] [CrossRef]
- Malipatil, M. Industry Biosecurity Plan for the Grains Industry. Threat Specific Contingency Plan Barley Stem Gall Midge. Mayetiola hordei; Plant Health Australia: Canberra, Australia, 2008; p. 23. [Google Scholar]
- Lhaloui, S.; Bouhssini, M.E.; Otmane, R.; Ouriniche, S.; Alami, A. Comparative Biology and Life Cycle of The Barley Stem Gall Midge and Hessian fly (Diptera: Cecidomyiidae) in Morocco. Rev. Maroc. Prot. Plantes 2016, 17–37. Available online: https://revues.imist.ma/index.php/RMPP/article/view/6420 (accessed on 12 March 2023).
- Parker, B.; El-Bouhssini, M.; Skinner, M. Field Guide: Insect Pests of Wheat and Barley in North Africa, West and Central Asia; ICARDA: Beirut, Lebanon, 2001; ISBN 978-92-9127-114-6. [Google Scholar]
- Hardie, J.; Minks, A.K. Pheromones of Non-Lepidopteran Insects Associated with Agricultural Plants; CABI Publishing: Wallingford, UK, 1999. [Google Scholar]
- Lhaloui, S.; Bouhssini, M.E.; Naserlhaq, N.; Amri, A.; Nachit, M.; Haddoury, J.E. Les Cecidomyies Des Cereales Au Maroc; INRA: Rabat, Morocco, 2005; 52p, ISBN 9981-1994-7-8. [Google Scholar]
- Schmid, R.B.; Knutson, A.; Giles, K.L.; McCornack, B.P. Hessian Fly (Diptera: Cecidomyiidae) Biology and Management in Wheat. J. Integr. Pest Manag. 2018, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Douglas, A.E. Lessons from Studying Insect Symbioses. Cell Host Microbe 2011, 10, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Bourtzis, K.; Miller, T.A. (Eds.) Insect symbiosis; Contemporary Topics in Entomology Series; CRC Press: Boca Raton, FL, USA, 2003; ISBN 978-0-8493-1286-1. [Google Scholar]
- Sanchez-Contreras, M.; Vlisidou, I. The Diversity of Insect-bacteria Interactions and its Applications for Disease Control. Biotechnol. Genet. Eng. Rev. 2008, 25, 203–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, P. Biology bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu. Rev. Microbiol. 2005, 59, 155–189. [Google Scholar] [CrossRef]
- Zchori-Fein, E.; Bourtzis, K. Manipulative Tenants: Bacteria Associated with Arthropods; CRC Press: Boca Raton, FL, USA, 2011; ISBN 978-1-4398-2749-9. [Google Scholar]
- Buchner, P.; Buchner, P. Endosymbiosis of animals with plant microorganisms; Interscience: New York, NY, USA, 1965. [Google Scholar]
- Douglas, A.E. Symbiotic microorganisms: Untapped resources for insect pest control. Trends Biotechnol. 2007, 25, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Hertig, M.; Wolbach, S.B. Studies on Rickettsia-Like Micro-Organisms in Insects. J. Med. Res. 1924, 44, 329–374.7. [Google Scholar] [PubMed]
- Lefoulon, E.; Clark, T.; Guerrero, R.; Cañizales, I.; Cardenas-Callirgos, J.M.; Junker, K.; Vallarino-Lhermitte, N.; Makepeace, B.L.; Darby, A.C.; Foster, J.M.; et al. Diminutive, degraded but dissimilar: Wolbachia genomes from filarial nematodes do not conform to a single paradigm. Microb. Genom. 2020, 6, e000487. [Google Scholar] [CrossRef]
- Fenn, K.; Conlon, C.; Jones, M.; Quail, M.A.; Holroyd, N.E.; Parkhill, J.; Blaxter, M. Phylogenetic Relationships of the Wolbachia of Nematodes and Arthropods. PLoS Pathog. 2006, 2, e94. [Google Scholar] [CrossRef] [Green Version]
- Lefoulon, E.; Clark, T.; Borveto, F.; Perriat-Sanguinet, M.; Moulia, C.; Slatko, B.E.; Gavotte, L. Pseudoscorpion Wolbachia symbionts: Diversity and evidence for a new supergroup S. BMC Microbiol 2020, 20, 188. [Google Scholar] [CrossRef] [PubMed]
- Rowley, S.M.; Raven, R.J.; McGraw, E.A. Wolbachia pipientis in Australian Spiders. Curr Microbiol 2004, 49, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Baldo, L.; Werren, J.H. Revisiting Wolbachia Supergroup Typing Based on WSP: Spurious Lineages and Discordance with MLST. Curr. Microbiol. 2007, 55, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-H.; Jia, L.-Y.; Xiao, J.-H.; Huang, D.-W. Discovery of a new Wolbachia supergroup in cave spider species and the lateral transfer of phage WO among distant hosts. Infect. Genet. Evol. 2016, 41, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerth, M. Classification of Wolbachia (Alphaproteobacteria, Rickettsiales): No Evidence for a Distinct Supergroup in Cave Spiders. bioRxiv 2016. [Google Scholar] [CrossRef] [PubMed]
- Bourtzis, K. Wolbachia-Based Technologies for Insect Pest Population Control. In Transgenesis and the Management of Vector-Borne Disease; Aksoy, S., Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2008; pp. 104–113. ISBN 978-0-387-78225-6. [Google Scholar]
- Saridaki, A.; Bourtzis, K. Wolbachia: More than just a bug in insects genitals. Curr. Opin. Microbiol. 2010, 13, 67–72. [Google Scholar] [CrossRef]
- Vreysen, M.J.B.; Robinson, A.S.; Hendrichs, J. Area-Wide Control of Insect Pests: From Research to Field Implementation; Springer: Dordrecht, The Netherlands, 2007; ISBN 978-1-4020-6058-8. [Google Scholar]
- Correa, C.C.; Ballard, J.W.O. Wolbachia Associations with Insects: Winning or Losing Against a Master Manipulator. Front. Ecol. Evol. 2016, 3, 153. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Butler, S.; Sanchez, G.; Mateos, M. Male killing Spiroplasma protects Drosophila melanogaster against two parasitoid wasps. Heredity 2014, 112, 399–408. [Google Scholar] [CrossRef]
- Anbutsu, H.; Fukatsu, T. Spiroplasma as a model insect endosymbiont. Environ. Microbiol. Rep. 2011, 3, 144–153. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Chen, Y.-T.; Yang, K.; Hong, X.-Y. A review of prevalence and phylogeny of the bacterial symbiont Cardinium in mites (subclass: Acari). Syst. Appl. Acarol. 2016, 21, 978. [Google Scholar] [CrossRef] [Green Version]
- Zchori-Fein, E.; Perlman, S.J.; Kelly, S.E.; Katzir, N.; Hunter, M.S.Y. Characterization of a ‘Bacteroidetes’ symbiont in Encarsia wasps (Hymenoptera: Aphelinidae): Proposal of ‘Candidatus Cardinium hertigii’. Int. J. Syst. Evol. Microbiol. 2004, 54, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Ferree, P.M.; Avery, A.; Azpurua, J.; Wilkes, T.; Werren, J.H. A Bacterium Targets Maternally Inherited Centrosomes to Kill Males in Nasonia. Curr. Biol. 2008, 18, 1409–1414. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.P.; Coghlin, P.C.; Floate, K.D.; Perlman, S.J. The host range of the male-killing symbiont Arsenophonus nasoniae in filth fly parasitioids. J. Invertebr. Pathol. 2011, 106, 371–379. [Google Scholar] [CrossRef]
- Mezghani Khemakhem, M.; Makni, H.; Marrakchi, M. PCR-RFLP identification of mitochondrial markers of Mayetiola (Diptera: Cecidomyiidae) on cereals. Ann. Soc. Entomol. Fr. 2002, 38, 277–282. [Google Scholar]
- Bel Mokhtar, N.; Maurady, A.; Britel, M.R.; El Bouhssini, M.; Batargias, C.; Stathopoulou, P.; Asimakis, E.; Tsiamis, G. Detection of Wolbachia Infections in Natural and Laboratory Populations of the Moroccan Hessian Fly, Mayetiola destructor (Say). Insects 2020, 11, 340. [Google Scholar] [CrossRef]
- Bansal, R.; Hulbert, S.; Schemerhorn, B.; Reese, J.C.; Whitworth, R.J.; Stuart, J.J.; Chen, M.-S. Hessian Fly-Associated Bacteria: Transmission, Essentiality, and Composition. PLoS ONE 2011, 6, e23170. [Google Scholar] [CrossRef] [Green Version]
- Augustinos, A.A.; Santos-Garcia, D.; Dionyssopoulou, E.; Moreira, M.; Papapanagiotou, A.; Scarvelakis, M.; Doudoumis, V.; Ramos, S.; Aguiar, A.F.; Borges, P.A.V.; et al. Detection and Characterization of Wolbachia Infections in Natural Populations of Aphids: Is the Hidden Diversity Fully Unraveled? PLoS ONE 2011, 6, e28695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, J.L.; Bowen, H. PEG precipitation for selective removal of small DNA fragments. Focus 2003, 25, 18. [Google Scholar]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UNCROSS2: Identification of cross-talk in 16S rRNA OTU tables. bioRxiv 2018. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Hollander, M.; Wolfe, D.A.; Chicken, E. Nonparametric Statistical Methods, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; ISBN 978-0-470-38737-5. [Google Scholar]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef] [Green Version]
- Steele, J.A.; Countway, P.D.; Xia, L.; Vigil, P.D.; Beman, J.M.; Kim, D.Y.; Chow, C.-E.T.; Sachdeva, R.; Jones, A.C.; Schwalbach, M.S.; et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011, 5, 1414–1425. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Yang, Y. Phylogenetic Molecular Ecological Network of Soil Microbial Communities in Response to Elevated CO2. mBio 2011, 2, e00122-11. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Raes, J. CoNet app: Inference of biological association networks using Cytoscape. F1000Res 2016, 5, 1519. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akaike, H. Autoregressive model fitting for control. Ann. Inst. Stat. Math. 1971, 23, 163–180. [Google Scholar] [CrossRef]
- Comar, M.; D’Accolti, M.; Cason, C.; Soffritti, I.; Campisciano, G.; Lanzoni, L.; Bisi, M.; Volta, A.; Mazzacane, S.; Caselli, E. Introduction of NGS in Environmental Surveillance for Healthcare-Associated Infection Control. Microorganisms 2019, 7, 708. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.J. Permutation tests for univariate or multivariate analysis of variance and regression. Can. J. Fish. Aquat. Sci. 2001, 58, 626–639. [Google Scholar] [CrossRef]
- Vandekerckhove, T.T.M.; Watteyne, S.; Willems, A.; Swings, J.G.; Mertens, J.; Gillis, M. Phylogenetic analysis of the 16S rDNA of the cytoplasmic bacterium Wolbachia from the novel host Folsomia candida (Hexapoda, Collembola) and its implications for Wolbachial taxonomy. FEMS Microbiol. Lett. 1999, 180, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Carvajal, T.M.; Hashimoto, K.; Harnandika, R.K.; Amalin, D.M.; Watanabe, K. Detection of Wolbachia in field-collected Aedes aegypti mosquitoes in metropolitan Manila, Philippines. Parasites Vectors 2019, 12, 361. [Google Scholar] [CrossRef] [Green Version]
- Lo, N.; Casiraghi, M.; Salati, E.; Bazzocchi, C.; Bandi, C. How Many Wolbachia Supergroups Exist? Mol. Biol. Evol. 2002, 19, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Fanning, S.; Proos, S.; Jordan, K.; Srikumar, S. A Review on the Applications of Next Generation Sequencing Technologies as Applied to Food-Related Microbiome Studies. Front. Microbiol. 2017, 8, 1829. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.J.; Schemerhorn, B.J.; Shukle, R.H. A First Assessment of Mitochondrial DNA Variation and Geographic Distribution of Haplotypes in Hessian fly (Diptera: Cecidomyiidae). Ann. Entomol. Soc. Am. 2004, 97, 940–948. [Google Scholar] [CrossRef]
- Johnson, A.J.; Morton, P.K.; Schemerhorn, B.J.; Shukle, R.H. Use of a Nuclear Marker to Assess Population Structure in Hessian Fly (Diptera: Cecidomyiidae). Ann. Entomol. Soc. Am. 2011, 104, 666–674. [Google Scholar] [CrossRef]
- Ojha, A.; Sinha, D.K.; Padmakumari, A.P.; Bentur, J.S.; Nair, S. Bacterial Community Structure in the Asian Rice Gall Midge Reveals a Varied Microbiome Rich in Proteobacteria. Sci. Rep. 2017, 7, 9424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glowska, E.; Dragun-Damian, A.; Dabert, M.; Gerth, M. New Wolbachia supergroups detected in quill mites (Acari: Syringophilidae). Infect. Genet. Evol. 2015, 30, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Zug, R.; Hammerstein, P. Still a Host of Hosts for Wolbachia: Analysis of Recent Data Suggests That 40% of Terrestrial Arthropod Species Are Infected. PLoS ONE 2012, 7, e38544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilgenboecker, K.; Hammerstein, P.; Schlattmann, P.; Telschow, A.; Werren, J.H. How many species are infected with Wolbachia?—A statistical analysis of current data: Wolbachia infection rates. FEMS Microbiol. Lett. 2008, 281, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyaprakash, A.; Hoy, M.A. Long PCR improves Wolbachia DNA amplification: Wsp sequences found in 76% of sixty-three arthropod species. Insect Mol. Biol. 2000, 9, 393–405. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, Z.-R.; Song, Y.; Liu, H.-Y.; Li, Z.-X. Distribution and diversity of Wolbachia in different populations of the wheat aphid Sitobion miscanthi (Hemiptera: Aphididae) in China. Eur. J. Entomol. 2009, 106, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Zytynska, S.E.; Weisser, W.W. The natural occurrence of secondary bacterial symbionts in aphids: Aphid secondary symbionts. Ecol. Entomol. 2016, 41, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Mateos, M.; Martinez Montoya, H.; Lanzavecchia, S.B.; Conte, C.; Guillén, K.; Morán-Aceves, B.M.; Toledo, J.; Liedo, P.; Asimakis, E.D.; Doudoumis, V.; et al. Wolbachia pipientis Associated With Tephritid Fruit Fly Pests: From Basic Research to Applications. Front. Microbiol. 2020, 11, 1080. [Google Scholar] [CrossRef]
- Asimakis, E.D.; Doudoumis, V.; Hadapad, A.B.; Hire, R.S.; Batargias, C.; Niu, C.; Khan, M.; Bourtzis, K.; Tsiamis, G. Detection and characterization of bacterial endosymbionts in Southeast Asian tephritid fruit fly populations. BMC Microbiol. 2019, 19, 290. [Google Scholar] [CrossRef]
- Sun, X.; Cui, L.; Li, Z. Diversity and Phylogeny of Wolbachia Infecting Bactrocera dorsalis (Diptera: Tephritidae) Populations from China. Environ. Entomol. 2007, 36, 1283–1289. [Google Scholar] [CrossRef]
- González Villa, T.; Viñas, M. (Eds.) Horizontal Gene Transfer: Breakting Borders between Living Kingdoms; Springer: Cham, Switzerland, 2019; ISBN 978-3-030-21862-1. [Google Scholar]
- Kondo, N.; Nikoh, N.; Ijichi, N.; Shimada, M.; Fukatsu, T. Genome fragment of Wolbachia endosymbiont transferred to X chromosome of host insect. Proc. Natl. Acad. Sci. USA 2002, 99, 14280–14285. [Google Scholar] [CrossRef] [Green Version]
- Bordenstein, S.R.; Bordenstein, S.R. Temperature Affects the Tripartite Interactions between Bacteriophage WO, Wolbachia, and Cytoplasmic Incompatibility. PLoS ONE 2011, 6, e29106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.Z.; De Barro, P.J.; Ren, S.-X.; Greeff, J.M.; Qiu, B.-L. Evidence for Horizontal Transmission of Secondary Endosymbionts in the Bemisia tabaci Cryptic Species Complex. PLoS ONE 2013, 8, e53084. [Google Scholar] [CrossRef] [PubMed]
- Huigens, M.E.; de Almeida, R.P.; Boons, P.A.H.; Luck, R.F.; Stouthamer, R. Natural interspecific and intraspecific horizontal transfer of parthenogenesis–inducing Wolbachia in Trichogramma wasps. Proc. R. Soc. Lond. B 2004, 271, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.Z.; Li, S.-J.; Xue, X.; Yin, X.-J.; Ren, S.-X.; Jiggins, F.M.; Greeff, J.M.; Qiu, B.-L. The Intracellular Bacterium Wolbachia Uses Parasitoid Wasps as Phoretic Vectors for Efficient Horizontal Transmission. PLoS Pathog. 2015, 11, e1004672. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-J.; Ahmed, M.Z.; Lv, N.; Shi, P.-Q.; Wang, X.-M.; Huang, J.-L.; Qiu, B.-L. Plantmediated horizontal transmission of Wolbachia between whiteflies. ISME J. 2017, 11, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.A.; Ross, P.A.; Rašić, G. Wolbachia strains for disease control: Ecological and evolutionary considerations. Evol. Appl. 2015, 8, 751–768. [Google Scholar] [CrossRef]
- Hughes, G.L.; Rasgon, J.L. Transinfection: A method to investigate Wolbachia-host interactions and control arthropod-borne disease: Transinfection of arthropods. Insect Mol. Biol. 2014, 23, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.L.; Barton, N.H.; Rašić, G.; Turley, A.P.; Montgomery, B.L.; Iturbe-Ormaetxe, I.; Cook, P.E.; Ryan, P.A.; Ritchie, S.A.; Hoffmann, A.A.; et al. Local introduction and heterogeneous spatial spread of dengue-suppressing Wolbachia through an urban population of Aedes aegypti. PLoS Biol. 2017, 15, e2001894. [Google Scholar] [CrossRef]
- McMeniman, C.J.; Lane, R.V.; Cass, B.N.; Fong, A.W.C.; Sidhu, M.; Wang, Y.-F.; O’Neill, S.L. Stable Introduction of a Life-Shortening Wolbachia Infection into the Mosquito Aedes aegypti. Science 2009, 323, 141–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, A.A.; Montgomery, B.L.; Popovici, J.; Iturbe-Ormaetxe, I.; Johnson, P.H.; Muzzi, F.; Greenfield, M.; Durkan, M.; Leong, Y.S.; Dong, Y.; et al. Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature 2011, 476, 454–457. [Google Scholar] [CrossRef]
- Ant, T.H.; Herd, C.; Louis, F.; Failloux, A.B.; Sinkins, S.P. Wolbachia transinfections in Culex quinquefasciatus generate cytoplasmic incompatibility. Insect Mol. Biol. 2020, 29, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolaki, A.; Livadaras, I.; Saridaki, A.; Chrysargyris, A.; Savakis, C.; Bourtzis, K. Transinfection of the olive fruit fly Bactrocera oleae with Wolbachia: Towards a symbiont-based population control strategy: Transinfection of Bactrocera oleae with Wolbachia. J. Appl. Entomol. 2011, 135, 546–553. [Google Scholar] [CrossRef]
- Kyritsis, G.A.; Koskinioti, P.; Bourtzis, K.; Papadopoulos, N.T. Effect of Wolbachia Infection and Adult Food on the Sexual Signaling of Males of the Mediterranean Fruit Fly Ceratitis capitata. Insects 2022, 13, 737. [Google Scholar] [CrossRef]
- Sarakatsanou, A.; Diamantidis, A.D.; Papanastasiou, S.A.; Bourtzis, K.; Papadopoulos, N.T. Effects of Wolbachia on fitness of the Mediterranean fruit fly (Diptera: Tephritidae): Wolbachia affects the fitness of C. capitata. J. Appl. Entomol. 2011, 135, 554–563. [Google Scholar] [CrossRef]
- Zabalou, S.; Apostolaki, A.; Livadaras, I.; Franz, G.; Robinson, A.S.; Savakis, C.; Bourtzis, K. Incompatible insect technique: Incompatible males from a Ceratitis capitata genetic sexing strain. Entomol. Exp. Et Appl. 2009, 132, 232–240. [Google Scholar] [CrossRef]
- Zabalou, S.; Riegler, M.; Theodorakopoulou, M.; Stauffer, C.; Savakis, C.; Bourtzis, K. Wolbachia-induced cytoplasmic incompatibility as a means for insect pest population control. Proc. Natl. Acad. Sci. USA 2004, 101, 15042–15045. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-H.; Chen, Y.; Li, W.; Tang, G.-H.; Yang, Y.; Jiang, H.-B.; Dou, W.; Wang, J.-J. Diversity of Bacterial Communities in the Intestinal Tracts of Two Geographically Distant Populations of Bactrocera dorsalis (Diptera: Tephritidae). J. Econ. Entomol. 2018, 11, 2861–2868. [Google Scholar] [CrossRef]
- Liu, L.J.; Martinez-Sañudo, I.; Mazzon, L.; Prabhakar, C.S.; Girolami, V.; Deng, Y.L.; Dai, Y.; Li, Z.H. Bacterial communities associated with invasive populations of Bactrocera dorsalis (Diptera: Tephritidae) in China. Bull. Entomol. Res. 2016, 106, 718–728. [Google Scholar] [CrossRef]
- Asimakis, E.; Stathopoulou, P.; Sapounas, A.; Khaeso, K.; Batargias, C.; Khan, M.; Tsiamis, G. New Insights on the Zeugodacus cucurbitae (Coquillett) Bacteriome. Microorganisms 2021, 9, 659. [Google Scholar] [CrossRef] [PubMed]
- Yong, H.-S.; Song, S.-L.; Eamsobhana, P.; Pasartvit, A.; Lim, P.-E. Differential abundance and core members of the bacterial community associated with wild male Zeugodacus cucurbitae fruit flies (Insecta: Tephritidae) from three geographical regions of Southeast Asia. Mol. Biol. Rep. 2019, 46, 3765–3776. [Google Scholar] [CrossRef]
- Takemura, M. Genome Sequence of a New “Candidatus” Phylum “Dependentiae” Isolate from Chiba, Japan. Microbiol. Resour. Announc. 2022, 11, e01123-21. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.S.; Lombardo, M.-J.; Badger, J.H.; Edlund, A.; Novotny, M.; Yee-Greenbaum, J.; Vyahhi, N.; Hall, A.P.; Yang, Y.; Dupont, C.L.; et al. Candidate phylum TM6 genome recovered from a hospital sink biofilm provides genomic insights into this uncultivated phylum. Proc. Natl. Acad. Sci. USA 2013, 110, E2390–E2399. [Google Scholar] [CrossRef] [Green Version]
- Pagnier, I.; Yutin, N.; Croce, O.; Makarova, K.S.; Wolf, Y.I.; Benamar, S.; Raoult, D.; Koonin, E.V.; La Scola, B. Babela massiliensis, a representative of a widespread bacterial phylum with unusual adaptations to parasitism in amoebae. Biol. Direct 2015, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delafont, V.; Samba-Louaka, A.; Bouchon, D.; Moulin, L.; Héchard, Y. Shedding light on microbial dark matter: A TM6 bacterium as natural endosymbiont of a free-living amoeba: TM6 bacterium as endosymbiont of amoeba. Environ. Microbiol. Rep. 2015, 7, 970–978. [Google Scholar] [CrossRef]
- Deeg, C.M.; Zimmer, M.M.; George, E.E.; Husnik, F.; Keeling, P.J.; Suttle, C.A. Chromulinavorax destructans, a pathogen of microzooplankton that provides a window into the enigmatic candidate phylum Dependentiae. PLoS Pathog. 2019, 15, e1007801. [Google Scholar] [CrossRef] [Green Version]
- Yutin, N.; Galperin, M.Y. A genomic update on clostridial phylogeny: Gram-negative spore formers and other misplaced clostridia: Genomics update. Env. Microbiol 2013, 15, 2631–2641. [Google Scholar] [CrossRef]
- Marchandin, H.; Teyssier, C.; Campos, J.; Jean-Pierre, H.; Roger, F.; Gay, B.; Carlier, J.-P.; Jumas-Bilak, E. Negativicoccus succinicivorans gen. nov., sp. nov., isolated from human clinical samples, emended description of the family Veillonellaceae and description of Negativicutes classis nov., Selenomonadales ord. nov. and Acidaminococcaceae fam. nov. in the bacterial phylum Firmicutes. Int. J. Syst. Evol. Microbiol. 2010, 60, 1271–1279. [Google Scholar] [CrossRef]
- Campbell, C.; Adeolu, M.; Gupta, R.S. Genome-based taxonomic framework for the class Negativicutes: Division of the class Negativicutes into the orders Selenomonadales emend., Acidaminococcales ord. nov. and Veillonellales ord. nov. Int. J. Syst. Evol. Microbiol. 2015, 65, 3203–3215. [Google Scholar] [CrossRef]
- Beskrovnaya, P.; Fakih, D.; Morneau, I.; Hashimi, A.; Guadarrama Bello, D.; Xing, S.; Nanci, A.; Huan, T.; Tocheva, E.I. No Endospore Formation Confirmed in Members of the Phylum Proteobacteria. Appl Env. Microbiol. 2021, 87, e02312-20. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.K.C.; Hamilton, I.R. Lactate Metabolism by Veillonella parvula. J. Bacteriol. 1971, 105, 999–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.C.; Levy, S.B.; Jackson, R.W. Pseudomonas genomes: Diverse and adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crone, S.; Vives-Flórez, M.; Kvich, L.; Saunders, A.M.; Malone, M.; Nicolaisen, M.H.; Martínez-García, E.; Rojas-Acosta, C.; Catalina Gomez-Puerto, M.; Calum, H.; et al. The environmental occurrence of Pseudomonas aeruginosa. APMIS 2020, 128, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Mercado-Blanco, J.; Bakker, P.A.H.M. Interactions between plants and beneficial Pseudomonas spp.: Exploiting bacterial traits for crop protection. Antonie Leeuwenhoek 2007, 92, 367–389. [Google Scholar] [CrossRef]
- Xin, X.-F.; Kvitko, B.; He, S.Y. Pseudomonas syringae: What it takes to be a pathogen. Nat. Rev. Microbiol. 2018, 16, 316–328. [Google Scholar] [CrossRef]
- Huszczynski, S.M.; Lam, J.S.; Khursigara, C.M. The Role of Pseudomonas aeruginosa Lipopolysaccharide in Bacterial Pathogenesis and Physiology. Pathogens 2019, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Ramos, J.-L.; Goldberg, J.B.; Filloux, A. (Eds.) Pseudomonas; Springer: Dordrecht, The Netherlands, 2015; ISBN 978-94-017-9554-8. [Google Scholar]
- Kim, H.R.; Lee, H.M.; Yu, H.C.; Jeon, E.; Lee, S.; Li, J.; Kim, D.-H. Biodegradation of Polystyrene by Pseudomonas sp. Isolated from the Gut of Superworms (Larvae of Zophobas atratus). Environ. Sci. Technol. 2020, 54, 6987–6996. [Google Scholar] [CrossRef]
- Looney, W.J.; Narita, M.; Mühlemann, K. Stenotrophomonas maltophilia: An emerging opportunist human pathogen. Lancet Infect. Dis. 2009, 9, 312–323. [Google Scholar] [CrossRef]
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; van der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef]
- Berg, G.; Martinez, J.L. Friends or foes: Can we make a distinction between beneficial and harmful strains of the Stenotrophomonas maltophilia complex? Front. Microbiol. 2015, 6, 241. [Google Scholar] [CrossRef]
- Gouveia, C.; Asensi, M.D.; Zahner, V.; Rangel, E.F.; Oliveira, S.M.P. de Study on the bacterial midgut microbiota associated to different Brazilian populations of Lutzomyia longipalpis (Lutz & Neiva) (Diptera: Psychodidae). Neotrop. Entomol. 2008, 37, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Morales-Jiménez, J.; Zúñiga, G.; Villa-Tanaca, L.; Hernández-Rodríguez, C. Bacterial Community and Nitrogen Fixation in the Red Turpentine Beetle, Dendroctonus valens LeConte (Coleoptera: Curculionidae: Scolytinae). Microb. Ecol. 2009, 58, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Völksch, B.; Thon, S.; Jacobsen, I.D.; Gube, M. Polyphasic study of plant- and clinic-associated Pantoea agglomerans strains reveals indistinguishable virulence potential. Infect. Genet. Evol. 2009, 9, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Pusey, P.L.; Stockwell, V.O.; Reardon, C.L.; Smits, T.H.M.; Duffy, B. Antibiosis Activity of Pantoea agglomerans Biocontrol Strain E325 Against Erwinia amylovora on Apple Flower Stigmas. Phytopathology 2011, 101, 1234–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, A.; Kohchi, C.; Inagawa, H.; Nishizawa, T.; Soma, G.-I. Improvement of Allergic Dermatitis via Regulation of the Th1/Th2 Immune System Balance by Macrophages Activated with Lipopolysaccharide Derived from Pantoea agglomerans (IP-PA1). Anticancer Res. 2009, 29, 4867–4870. [Google Scholar] [PubMed]
- Hebishima, T.; Matsumoto, Y.; Watanabe, G.; Soma, G.; Kohchi, C.; Taya, K.; Hayashi, Y.; Hirota, Y. Oral Administration of Immunopotentiator from Pantoea agglomerans 1 (IP-PA1) Improves the Survival of B16 Melanoma-Inoculated Model Mice. Exp. Anim. 2011, 60, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Nakata, K.; Inagawa, H.; Soma, G.-I. Lipopolysaccharide IP-PA1 from Pantoea agglomerans Prevents Suppression of Macrophage Function in Stress-induced Diseases. Anticancer. Res. 2011, 7, 2437–2440. [Google Scholar]
- Johnson, K.B.; Stockwell, V.O.; McLaughlin, R.J.; Sugar, D.; Loper, J.E.; Roberts, R.G. Effect of antagonistic bacteria on establishment of honey bee-dispersed Erwinia amylovora in pear blossoms and on fire blight control. Phytopathology 1993, 83, 995–1002. [Google Scholar] [CrossRef]
- Johnson, K.B.; Stockwell, V.O.; Sawyer, T.L.; Sugar, D. Assessment of Environmental Factors Influencing Growth and Spread of Pantoea agglomerans on and Among Blossoms of Pear and Apple. Phytopathology 2000, 90, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.B.; Stockwell, V. Management of fire blight: A case study in microbial ecology. Annu. Rev. Phytopathol. 1998, 36, 227–248. [Google Scholar] [CrossRef] [PubMed]
- Toju, H.; Fukatsu, T. Diversity and infection prevalence of endosymbionts in natural populations of the chestnut weevil: Relevance of local climate and host plants: Endosymbionts in weevil populations. Mol. Ecol. 2011, 20, 853–868. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, R.K.; Fast, E.M.; Guo, R.; Schultz, M.J.; Vaisman, N.; Ortiz, L.; Bybee, J.; Slatko, B.E.; Frydman, H.M. The Gut Commensal Microbiome of Drosophila melanogaster Is Modified by the Endosymbiont Wolbachia. mSphere 2017, 2, e00287-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Location | Coordinates | Number of Insects | |||||
|---|---|---|---|---|---|---|---|---|
| Altitude | Latitude | Longitude | Temperature | Larvae | Male | Female | ||
| Abda | Ras El Ain | 183 | 32.16898 | −8.55100 | 23 °C | 8 | - | - |
| Khatazakane | 58 | 32.17595 | −9.10573 | 12 °C | 10 | - | - | |
| Rabat | Tiflet | 333 | 33.53909 | −6.21687 | 16 °C | 10 | - | - |
| SidiAllal Bahraoui | 166 | 34.00966 | −6.33159 | 16 °C | - | 11 | 11 | |
| Marchouch | 391 | 33.60060 | −6.710450 | 17 °C | 2 | - | - | |
| Rommani | 434 | 33.557.628 | −6.583154 | 19 °C | 10 | - | - | |
| Fes-Meknes | Iqaddar | 565 | 33.58706 | −5.35273 | 18 °C | 10 | - | - |
| Majjate | 706 | 33.780093 | −5.490483 | 19 °C | 20 | 11 | 11 | |
| Doukkala | Chaibate | 99 | 33.01083 | −8.29074 | 20 °C | 28 | - | - |
| Oulad Hamdane | 82 | 33.11217 | −8.23345 | 21 °C | 10 | - | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Remmal, I.; Bel Mokhtar, N.; Maurady, A.; Reda Britel, M.; El Fakhouri, K.; Asimakis, E.; Tsiamis, G.; Stathopoulou, P. Characterization of the Bacterial Microbiome in Natural Populations of Barley Stem Gall Midge, Mayetiola hordei, in Morocco. Microorganisms 2023, 11, 797. https://doi.org/10.3390/microorganisms11030797
Remmal I, Bel Mokhtar N, Maurady A, Reda Britel M, El Fakhouri K, Asimakis E, Tsiamis G, Stathopoulou P. Characterization of the Bacterial Microbiome in Natural Populations of Barley Stem Gall Midge, Mayetiola hordei, in Morocco. Microorganisms. 2023; 11(3):797. https://doi.org/10.3390/microorganisms11030797
Chicago/Turabian StyleRemmal, Imane, Naima Bel Mokhtar, Amal Maurady, Mohammed Reda Britel, Karim El Fakhouri, Elias Asimakis, George Tsiamis, and Panagiota Stathopoulou. 2023. "Characterization of the Bacterial Microbiome in Natural Populations of Barley Stem Gall Midge, Mayetiola hordei, in Morocco" Microorganisms 11, no. 3: 797. https://doi.org/10.3390/microorganisms11030797