
Metagenomes from Coastal Sediments of Kuwait: Insights into the Microbiome, Metabolic Functions and Resistome

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Methods
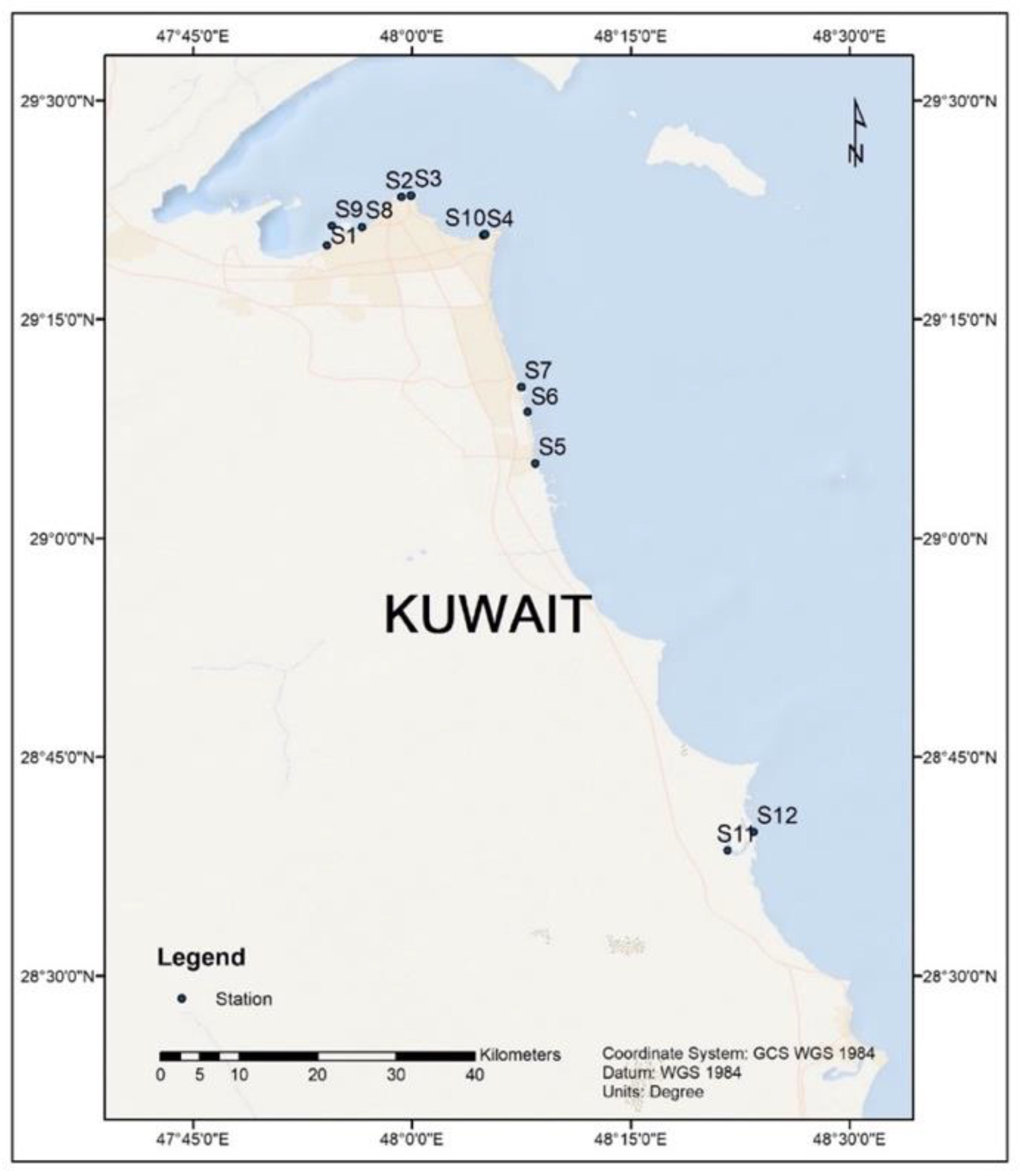
2.1. Sample Collection and DNA Extraction
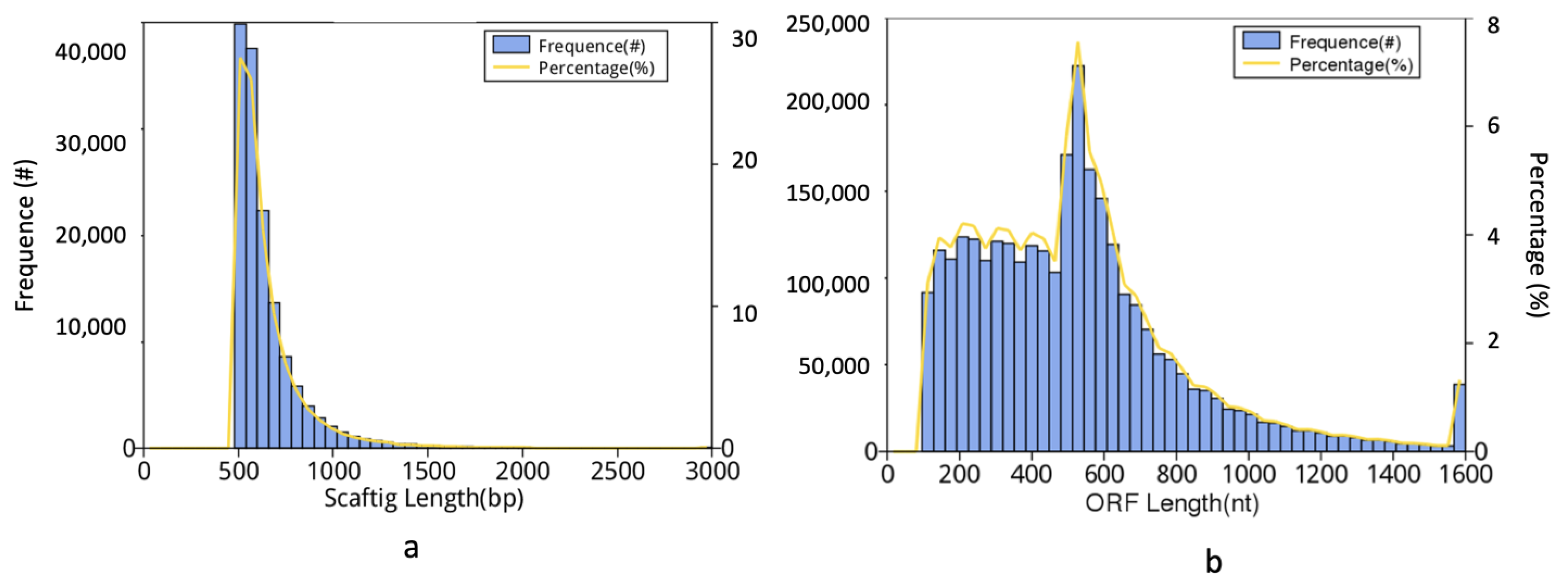
2.2. Metagenomic Sequencing
2.3. Functional Annotation
2.4. Statistical Analysis
3. Results
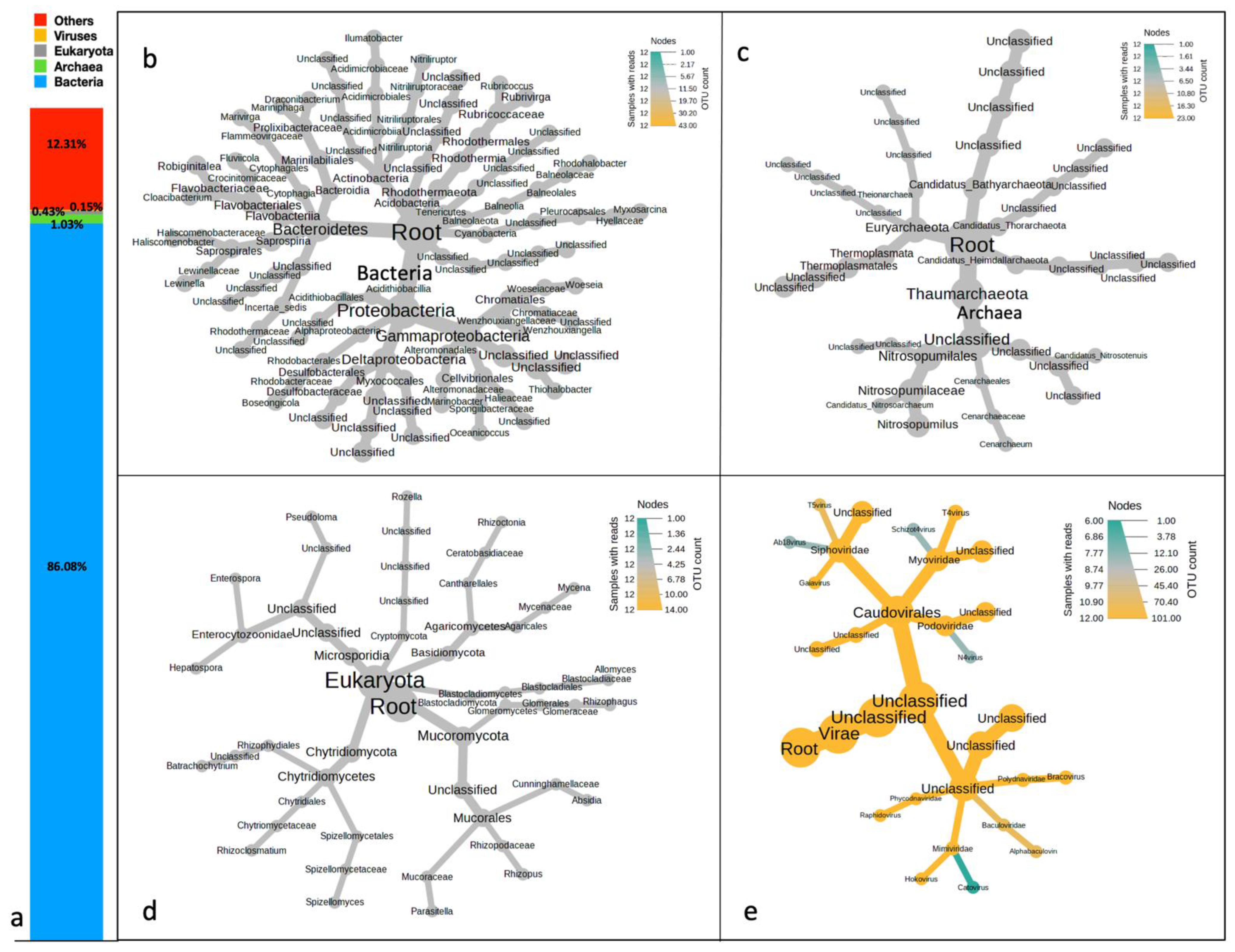
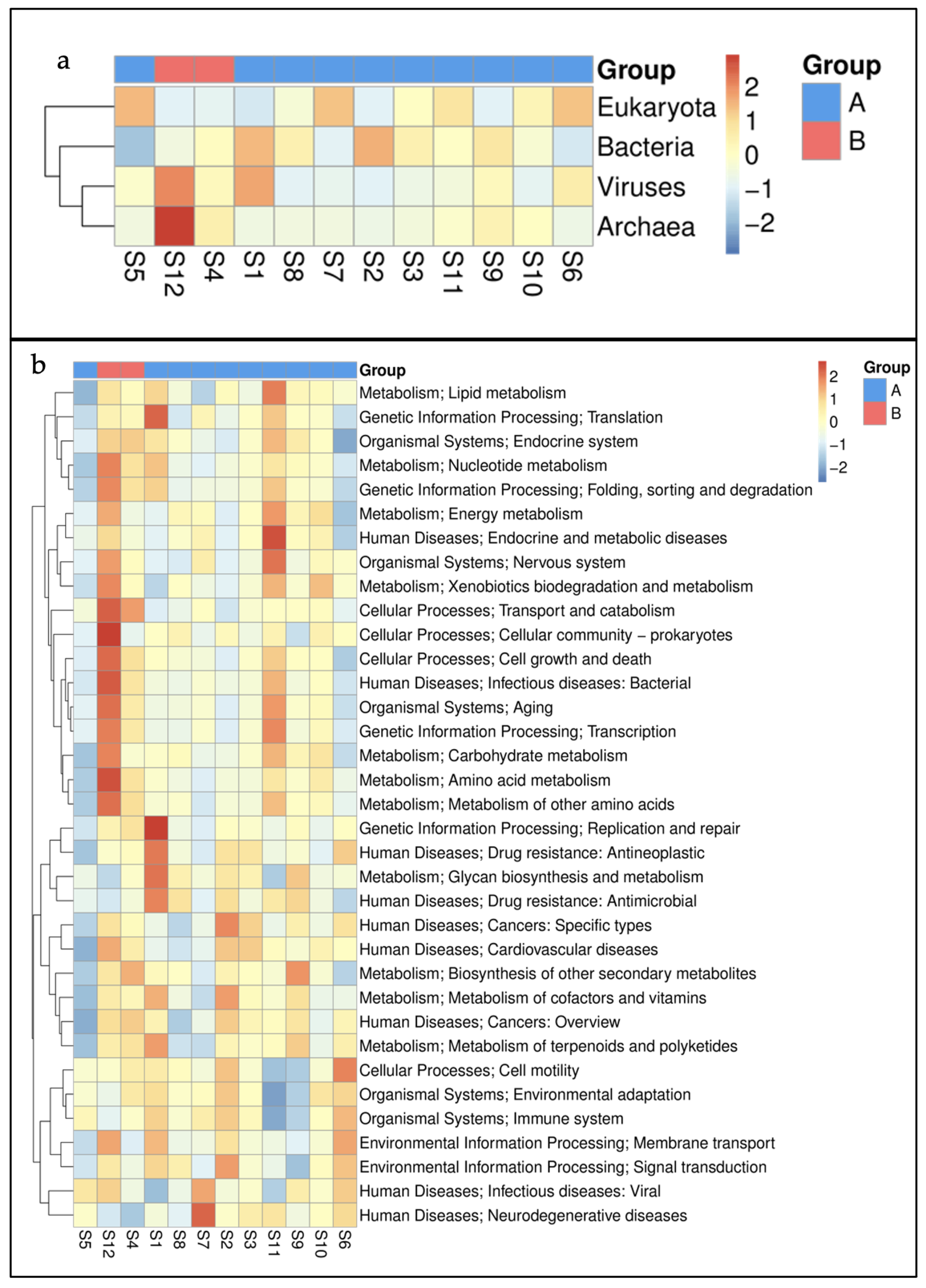
3.1. Taxonomic Profiling
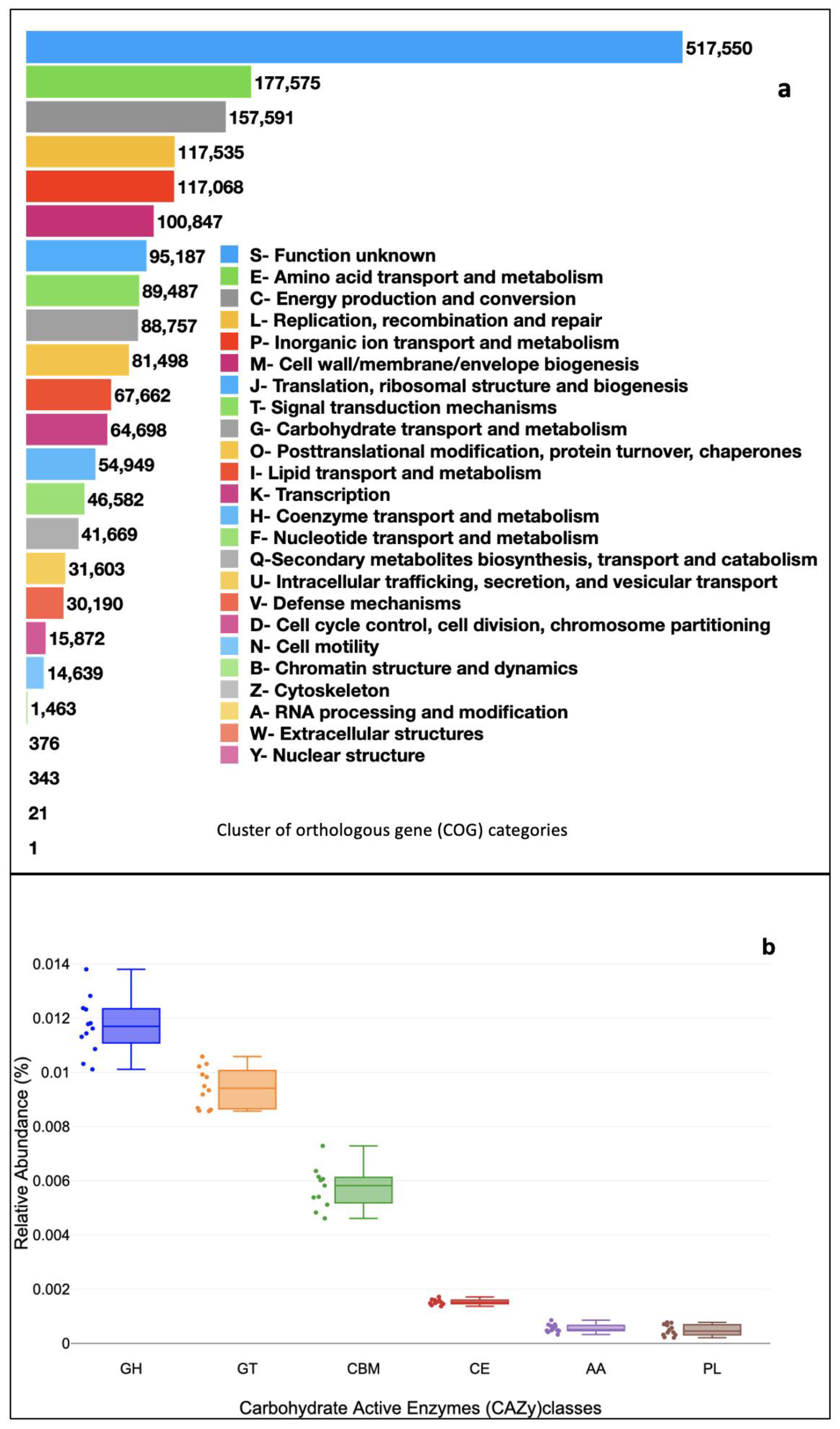
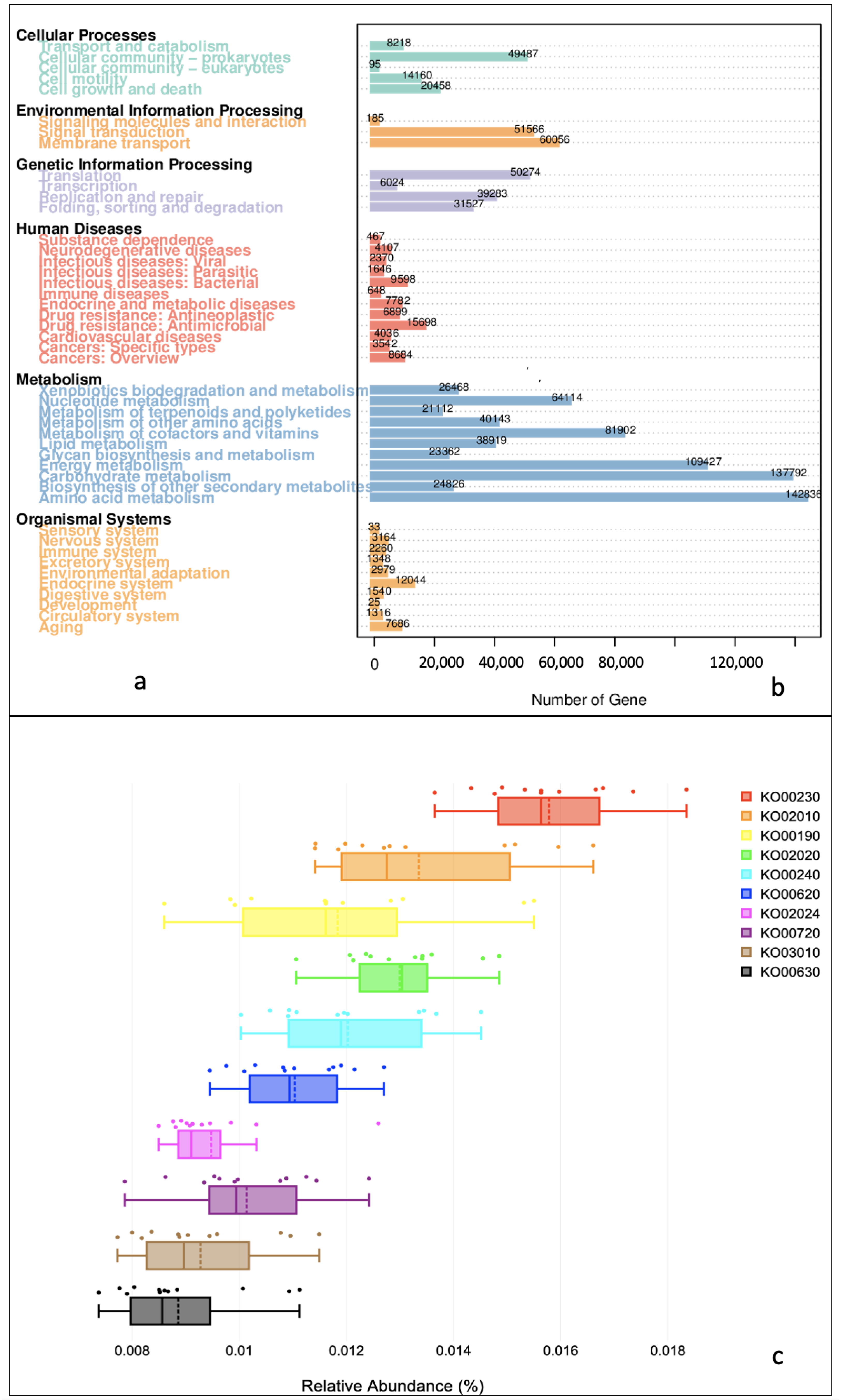
3.2. Functional Profiles
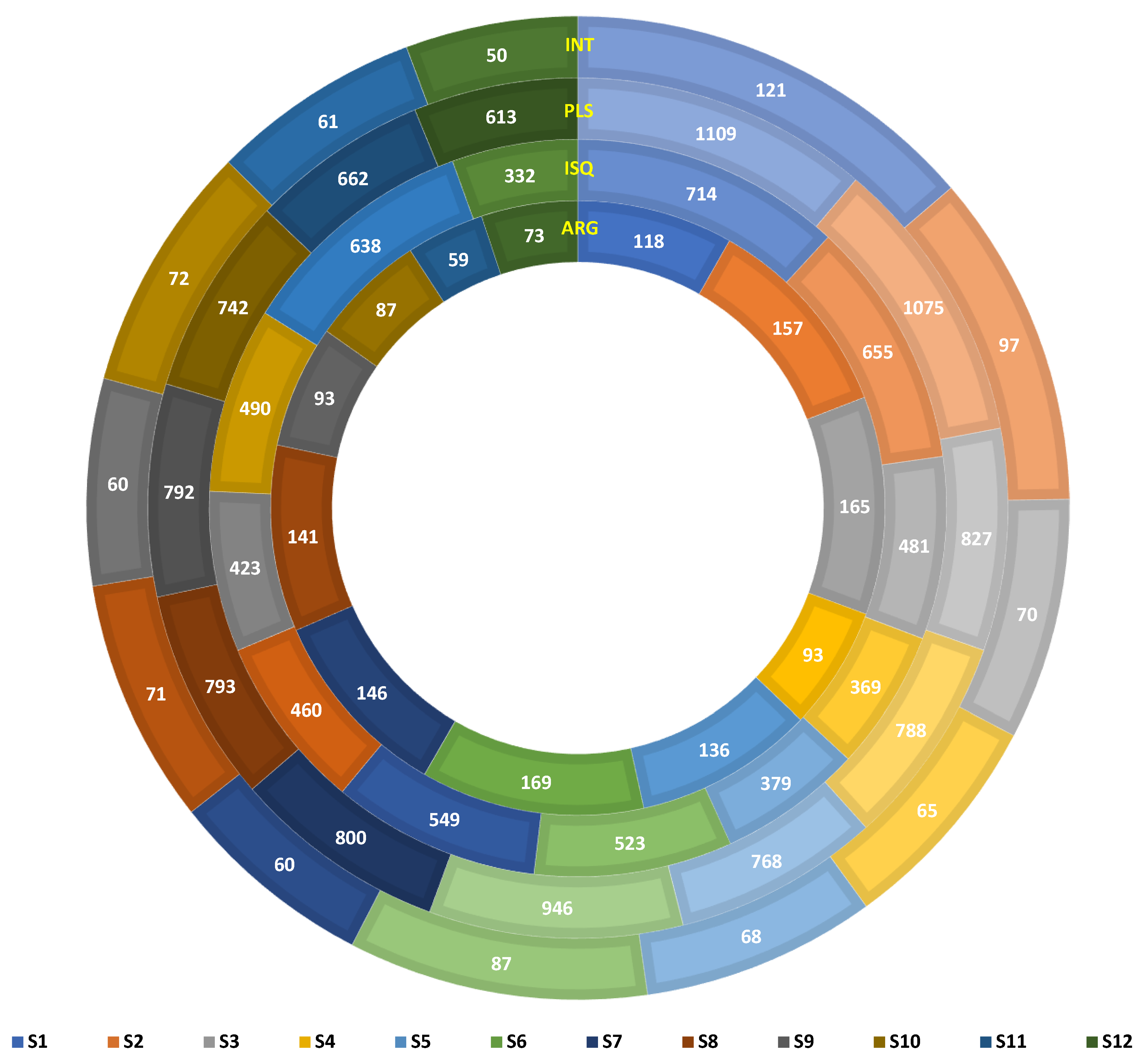
3.3. Resistome and Mobilome Profiling
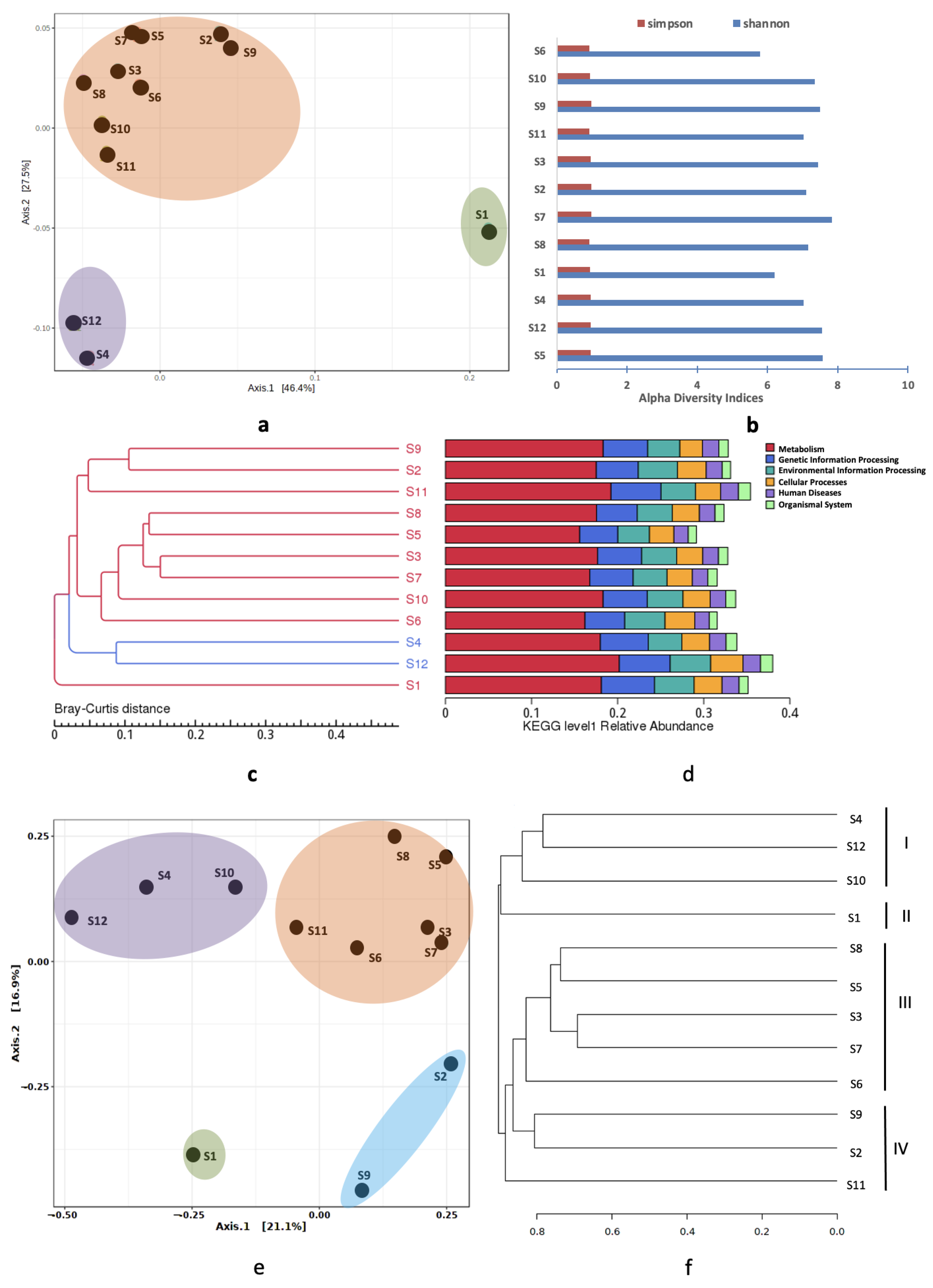
3.4. Spatial Variations
3.4.1. Alpha Diversity Analysis
3.4.2. Beta Diversity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balasubramanian, V.K.; Maran, M.I.J.; Ramteke, D.; Vijaykumar, D.S.; Rajendran, A.K.; Ramachandran, P.; Ramachandran, R. Environmental DNA reveals aquatic biodiversity of an urban backwater area, southeast coast of India. Mar. Pollut. Bull. 2021, 171, 112786. [Google Scholar] [CrossRef]
- Chen, J.; McIlroy, S.E.; Archana, A.; Baker, D.M.; Panagiotou, G. A pollution gradient contributes to the taxonomic, functional, and resistome diversity of microbial communities in marine sediments. Microbiome 2019, 7, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, N.M.; Hess, M.; Bouskill, N.J.; Mason, O.U.; Jansson, J.K.; Gilbert, J.A. The microbial nitrogen cycling potential is impacted by polyaromatic hydrocarbon pollution of marine sediments. Front. Microbiol. 2014, 5, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Yang, Y.; Liang, X.; Yu, K.; Zhang, T.; Li, X. Metagenomic profiles of antibiotic resistance genes (ARGs) between human impacted estuary and deep ocean sediments. Environ. Sci. Technol. 2013, 47, 12753–12760. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, J.D.; Cantão, M.E.; Cesar, D.E.; Nicolás, M.F.; Diniz, C.G.; Silva, V.L.; Vasconcelos, A.T.; Coelho, C.M. Comparative metagenome of a stream impacted by the urbanization phenomenon. Braz. J. Microbiol. 2016, 47, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Wu, R. Marine pollution in Hong Kong: A review. Asian Mar. Biol. 1988, 5, 1–23. [Google Scholar]
- Abreo, N.A.S.; Macusi, E.D.; Cuenca, G.C.; Ranara, C.T.B.; Andam, M.B.; Cardona, L.; Arabejo, G.F.P. Nutrient enrichment, sedimentation, heavy metals and plastic pollution in the marine environment and its implications on Philippine marine biodiversity: A Review. IAMURE Int. J. Ecol. Conserv. 2015, 15, 111–167. [Google Scholar] [CrossRef]
- WHO. List of bacteria for Which New Antibiotics Are Urgently Needed; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- WHO. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Xu, M.; Zhang, Q.; Xia, C.; Zhong, Y.; Sun, G.; Guo, J.; Yuan, T.; Zhou, J.; He, Z. Elevated nitrate enriches microbial functional genes for potential bioremediation of complexly contaminated sediments. ISME J. 2014, 8, 1932–1944. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, L.D.; Sørensen, S.J. The effect of longterm exposure to mercury on the bacterial community in marine sediment. Curr. Microbiol. 1998, 36, 291–297. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Y.; Xiang, S.-Z.; Yan, Y.; Yang, R.; Lin, M.-P.; Wang, X.-M.; Xue, Y.-L.; Guan, X.-Y. Sediment microbial communities and their potential role as environmental pollution indicators in Xuande Atoll, South China Sea. Front. Microbiol. 2020, 11, 1011. [Google Scholar] [CrossRef]
- Cyriac, M.; Gireeshkumar, T.; Furtado, C.; Fathin, K.F.; Shameem, K.; Shaik, A.; Vignesh, E.; Nair, M.; Kocherla, M.; Balachandran, K. Distribution, contamination status and bioavailability of trace metals in surface sediments along the southwest coast of India. Mar. Pollut. Bull. 2021, 164, 112042. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, R.; Liu, H.; Wang, C.; Yin, X.; Zhang, M.; Fang, J.; Zhang, T.; Ma, L. Evidence for Long-Term Anthropogenic Pollution: The Hadal Trench as a Depository and Indicator for Dissemination of Antibiotic Resistance Genes. Environ. Sci. Technol. 2021, 55, 15136–15148. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yan, L.; Yang, Y.; Zhou, H.; Cao, Y.; Wang, S.; Xue, B.; Li, C.; Zhao, C.; Zhang, X. The Occurrence and Distribution Pattern of Antibiotic Resistance Genes and Bacterial Community in the Ili River. Front. Environ. Sci. 2022, 10, 212. [Google Scholar] [CrossRef]
- Chen, Y.-r.; Guo, X.-p.; Niu, Z.-s.; Lu, D.-p.; Sun, X.-l.; Zhao, S.; Hou, L.-j.; Liu, M.; Yang, Y. Antibiotic resistance genes (ARGs) and their associated environmental factors in the Yangtze Estuary, China: From inlet to outlet. Mar. Pollut. Bull. 2020, 158, 111360. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-p.; Zhao, S.; Chen, Y.-r.; Yang, J.; Hou, L.-j.; Liu, M.; Yang, Y. Antibiotic resistance genes in sediments of the Yangtze Estuary: From 2007 to 2019. Sci. Total Environ. 2020, 744, 140713. [Google Scholar] [CrossRef]
- Mootapally, C.; Nathani, N.M.; Poriya, P.; Beleem, I.; Dabhi, J.C.; Gadhvi, I.R.; Joshi, C.G. Antibiotic Resistome Biomarkers associated to the pelagic Sediments of the Gulfs of Kathiawar peninsula and Arabian Sea. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Imchen, M.; Kumavath, R. Shotgun metagenomics reveals a heterogeneous prokaryotic community and a wide array of antibiotic resistance genes in mangrove sediment. FEMS Microbiol. Ecol. 2020, 96, fiaa173. [Google Scholar] [CrossRef]
- Catania, V.; Cappello, S.; Di Giorgi, V.; Santisi, S.; Di Maria, R.; Mazzola, A.; Vizzini, S.; Quatrini, P. Microbial communities of polluted sub-surface marine sediments. Mar. Pollut. Bull. 2018, 131, 396–406. [Google Scholar] [CrossRef]
- Ogaki, M.B.; Pinto, O.H.B.; Vieira, R.; Neto, A.A.; Convey, P.; Carvalho-Silva, M.; Rosa, C.A.; Câmara, P.E.; Rosa, L.H. Fungi present in Antarctic deep-sea sediments assessed using DNA metabarcoding. Microb. Ecol. 2021, 82, 157–164. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, W.; Liu, Y.; Cai, M.; Luo, Z.; Li, M. Metagenomics reveals microbial diversity and metabolic potentials of seawater and surface sediment from a hadal biosphere at the Yap Trench. Front. Microbiol. 2018, 9, 2402. [Google Scholar] [CrossRef] [Green Version]
- Cabello-Yeves, P.J.; Callieri, C.; Picazo, A.; Mehrshad, M.; Haro-Moreno, J.M.; Roda-Garcia, J.J.; Dzhembekova, N.; Slabakova, V.; Slabakova, N.; Moncheva, S.; et al. The microbiome of the Black Sea water column analyzed by shotgun and genome centric metagenomics. Environ. Microbiome 2021, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yan, Z.; Wang, P.; Zheng, X.; Fan, J. Comparative metagenomics reveals the microbial diversity and metabolic potentials in the sediments and surrounding seawaters of Qinhuangdao mariculture area. PLoS ONE 2020, 15, e0234128. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Wang, F.; Geng, H.; Liu, H.; Pu, S.; Tian, Z.; Chen, H.; Zhou, B.; Yuan, R.; Yao, J. Integrating high-throughput sequencing and metagenome analysis to reveal the characteristic and resistance mechanism of microbial community in metal contaminated sediments. Sci. Total Environ. 2020, 707, 136116. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Al Ghadban, A.N.; Khabbaz, A. Localized hyper saline waters in Arabian Gulf from desalination activity-an example from South Kuwait. Environ. Monit. Assess. 2011, 181, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Aba, A.; Fowler, S.W.; Behbehani, M.; Ismaeel, A.; Al-Shammari, H.; Alboloushi, A.; Mietelski, J.W.; Al-Ghadban, A.; Al-Ghunaim, A.; et al. Radioactivity in the Kuwait marine environment--Baseline measurements and review. Mar. Pollut. Bull. 2015, 100, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Aba, A.; Behbehani, M.; Al-Ghadban, A.N.; Al-Zekri, W.; Al-Shammari, H. Plutonium and cesium baseline concentrations in seawater from northern Arabian Gulf. Mar. Pollut. Bull. 2017, 120, 396–400. [Google Scholar] [CrossRef]
- Uddin, S.; Behbehani, M.; Aba, A.; Al Ghadban, A.N. Naturally Occurring Radioactive Material (NORM) in seawater of the northern Arabian Gulf—Baseline measurements. Mar. Pollut. Bull. 2017, 123, 365–372. [Google Scholar] [CrossRef]
- Alsarawi, H.A. Developing an Integrated Strategy for the Assessment of Hazardous Substances in Kuwait’s Marine Environment; University of Plymouth: Plymouth, UK, 2017. [Google Scholar]
- Al-Sarawi, H.A.; Jha, A.N.; Al-Sarawi, M.A.; Lyons, B.P. Historic and contemporary contamination in the marine environment of Kuwait: An overview. Mar. Pollut. Bull. 2015, 100, 621–628. [Google Scholar] [CrossRef]
- Gevao, B.; Uddin, S.; Dupont, S. Baseline concentrations of pharmaceuticals in Kuwait’s coastal marine environment. Mar. Pollut. Bull. 2021, 173, 113040. [Google Scholar] [CrossRef]
- Gevao, B.; Uddin, S.; Krishnan, D.; Rajagopalan, S.; Habibi, N. Antibiotics in Wastewater: Baseline of the Influent and Effluent Streams in Kuwait. Toxics 2022, 10, 174. [Google Scholar] [CrossRef]
- Al-Sarawi, H.A.; Jha, A.N.; Baker-Austin, C.; Al-Sarawi, M.A.; Lyons, B.P. Baseline screening for the presence of antimicrobial resistance in E. coli isolated from Kuwait’s marine environment. Mar. Pollut. Bull. 2018, 129, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarawi, H.A.; Najem, A.B.; Lyons, B.P.; Uddin, S.; Al-Sarawi, M.A. Antimicrobial Resistance in Escherichia coli Isolated from Marine Sediment Samples from Kuwait Bay. Sustainability 2022, 14, 11325. [Google Scholar] [CrossRef]
- Habibi, N.; Uddin, S.; Lyons, B.; Al-Sarawi, H.A.; Behbehani, M.; Shajan, A.; Razzack, N.A.; Zakir, F.; Alam, F. Antibiotic Resistance Genes Associated with Marine Surface Sediments: A Baseline from the Shores of Kuwait. Sustainability 2022, 14, 8029. [Google Scholar] [CrossRef]
- Devlin, M.; Le Quesne, W.J.; Lyons, B.P. The marine environment of Kuwait—Emerging issues in a rapidly changing environment. Mar. Pollut. Bull. 2015, 100, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Le Quesne, W.J.; Baker-Austin, C.; Verner-Jeffreys, D.W.; Al-Sarawi, H.A.; Balkhy, H.H.; Lyons, B.P. Antimicrobial resistance in the Gulf Cooperation Council region: A proposed framework to assess threats, impacts and mitigation measures associated with AMR in the marine and aquatic environment. Environ. Int. 2018, 121, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.W.; Habibi, N.; Shaheed, F.; Mustafa, A.S. Draft genome sequences of five clinical strains of Brucella melitensis isolated from patients residing in Kuwait. Genome Announc. 2016, 4, e01144-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Available online. Retrieved May 2010, 17, 2018. [Google Scholar]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, Z.S.; Sharpton, T.J.; Grünwald, N.J. MetacodeR: An R package for manipulation and heat tree visualization of community taxonomic data from metabarcoding. bioRxiv 2016, 071019. [Google Scholar] [CrossRef] [Green Version]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Drula, E.; Garron, M.-L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.D. Rarefaction, alpha diversity, and statistics. Front. Microbiol. 2019, 10, 2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doytchinov, V.V.; Dimov, S.G. Microbial community composition of the Antarctic ecosystems: Review of the bacteria, fungi, and archaea identified through an NGS-based metagenomics approach. Life 2022, 12, 916. [Google Scholar] [CrossRef]
- Nathani, N.M.; Mootapally, C.; Dave, B.P. Antibiotic resistance genes allied to the pelagic sediment microbiome in the Gulf of Khambhat and Arabian Sea. Sci. Total Environ. 2019, 653, 446–454. [Google Scholar] [CrossRef]
- Hoshino, T.; Doi, H.; Uramoto, G.-I.; Wörmer, L.; Adhikari, R.R.; Xiao, N.; Morono, Y.; D’Hondt, S.; Hinrichs, K.-U.; Inagaki, F. Global diversity of microbial communities in marine sediment. Proc. Natl. Acad. Sci. USA 2020, 117, 27587–27597. [Google Scholar] [CrossRef]
- Miettinen, H.; Bomberg, M.; Nyyssönen, M.; Reunamo, A.; Jørgensen, K.S.; Vikman, M. Oil degradation potential of microbial communities in water and sediment of Baltic Sea coastal area. PLoS ONE 2019, 14, e0218834. [Google Scholar] [CrossRef] [Green Version]
- Edgcomb, V.P.; Beaudoin, D.; Gast, R.; Biddle, J.F.; Teske, A. Marine subsurface eukaryotes: The fungal majority. Environ. Microbiol. 2011, 13, 172–183. [Google Scholar] [CrossRef]
- Pachiadaki, M.G.; Rédou, V.; Beaudoin, D.J.; Burgaud, G.; Edgcomb, V.P. Fungal and prokaryotic activities in the marine subsurface biosphere at Peru Margin and Canterbury Basin inferred from RNA-based analyses and microscopy. Front. Microbiol. 2016, 7, 846. [Google Scholar] [CrossRef] [Green Version]
- Breitbart, M.; Felts, B.; Kelley, S.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Diversity and population structure of a near–shore marine–sediment viral community. Proc. R. Soc. London Ser. B Biol. Sci. 2004, 271, 565–574. [Google Scholar] [CrossRef]
- Danovaro, R.; Corinaldesi, C.; Rastelli, E.; Anno, A.D. Towards a better quantitative assessment of the relevance of deep-sea viruses, Bacteria and Archaea in the functioning of the ocean seafloor. Aquat. Microb. Ecol. 2015, 75, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Al-Abdulghani, E.; El-Sammak, A.; Sarawi, M. Environmental assessment of Kuwait Bay: An integrated approach. J. Coast. Conserv. 2013, 17, 445–462. [Google Scholar] [CrossRef]
- Al-Gadhban, A.N.; Uddin, S.; Beg, M.U.; Al-Dousari, A.M.; Gevao, B. Ecological consequences of River Manipulation and Drainage of Mesopotamian Marshes on the Arabian Gulf Ecosystem: Investigations on Changes in Sedimentology and Environment Quality with Special Reference to Kuwait Bay; Kuwait Institute for Scientific Research: Kuwait City, Kuwait, 2008. [Google Scholar]
- Aba, A.; Uddin, S.; Bahbahani, M.; Al-Ghadban, A. Radiometric dating of sediment records in Kuwait’s marine area. J. Radioanal. Nucl. Chem. 2014, 301, 247–255. [Google Scholar] [CrossRef]
- Song, J.; Mujahid, A.; Lim, P.-T.; Samah, A.A.; Quack, B.; Pfeilsticker, K.; Tang, S.-L.; Ivanova, E.; Müller, M. Shotgun metagenomic analysis of microbial communities in the surface waters of the Eastern South China Sea. Malays. J. Microbiol. 2017, 350–362. [Google Scholar]
- He, L.; Huang, X.; Zhang, G.; Yuan, L.; Shen, E.; Zhang, L.; Zhang, X.-H.; Zhang, T.; Tao, L.; Ju, F. Distinctive signatures of pathogenic and antibiotic resistant potentials in the hadal microbiome. Environ. Microbiome 2022, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Alqattan, M.E.; Gray, T.S. Marine Pollution in Kuwait and Its Impacts on Fish-Stock Decline in Kuwaiti Waters: Reviewing the Kuwaiti Government’s Policies and Practices. Front. Sustain. 2021, 2, 51. [Google Scholar] [CrossRef]
- Korlević, M.; Zucko, J.; Dragić, M.N.; Blažina, M.; Pustijanac, E.; Zeljko, T.V.; Gacesa, R.; Baranasic, D.; Starcevic, A.; Diminic, J. Bacterial diversity of polluted surface sediments in the northern Adriatic Sea. Syst. Appl. Microbiol. 2015, 38, 189–197. [Google Scholar] [CrossRef]
- Uddin, S.; Fowler, S.W.; Saeed, T. Microplastic particles in the Persian/Arabian Gulf–a review on sampling and identification. Mar. Pollut. Bull. 2020, 154, 111100. [Google Scholar] [CrossRef]
- Uddin, S.; Fowler, S.W.; Uddin, M.F.; Behbehani, M.; Naji, A. A review of microplastic distribution in sediment profiles. Mar. Pollut. Bull. 2021, 163, 111973. [Google Scholar] [CrossRef]
- Al-Ghadban, A.N.; Uddin, S.; Maltby, E.; Al Khabbaz, A.; Al Mutairi, A. Denitrification potential of the Northern Arabian Gulf—An experimental study. Environ. Monit. Assess. 2012, 184, 7103–7112. [Google Scholar] [CrossRef]
- Liu, W.; Zheng, X.; Dai, X.; Zhang, Z.; Zhang, W.; Xiao, T.; Huang, L. Isolation and Characterization of the First Temperate Virus Infecting Psychrobacillus from Marine Sediments. Viruses 2022, 14, 108. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cai, L.; Zhang, R. Co-existence of freshwater and marine T4-like myoviruses in a typical subtropical estuary. FEMS Microbiol. Ecol. 2017, 93, fix119. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Al-Ghadban, A.N.; Gevao, B.; Al-Shamroukh, D.; Al-Khabbaz, A. Estimation of Suspended Particulate Matter in Gulf using MODIS data. Aquat. Ecosyst. Health Manag. Soc. 2012, 15, 41–44. [Google Scholar] [CrossRef]
- Elkadiri, R.; Manche, C.; Sultan, M.; Al-Dousari, A.; Uddin, S.; Chouinard, K.; Abotalib, A.Z. Development of a Coupled Spatiotemporal Algal Bloom Model for Coastal Areas: A Remote Sensing and Data Mining-Based Approach. IEEE J. Sel. Top. Appl. Earth Obs. Remote Sens. 2016, 9, 5159–5171. [Google Scholar] [CrossRef]
- Habibi, N.; Uddin, S.; Bottein, M.-Y.D.; Faizuddin, M. Ciguatera in the Indian Ocean with Special Insights on the Arabian Sea and Adjacent Gulf and Seas: A Review. Toxins 2021, 13, 525. [Google Scholar] [CrossRef]
- Nowak, M.A.; Boerlijst, M.C.; Cooke, J.; Smith, J.M. Evolution of genetic redundancy. Nature 1997, 388, 167–171. [Google Scholar] [CrossRef]
- Orsi, W.D.; Richards, T.A.; Francis, W.R. Predicted microbial secretomes and their target substrates in marine sediment. Nat. Microbiol. 2018, 3, 32–37. [Google Scholar] [CrossRef]
- Biddle, J.F.; White, J.R.; Teske, A.P.; House, C.H. Metagenomics of the subsurface Brazos-Trinity Basin (IODP site 1320): Comparison with other sediment and pyrosequenced metagenomes. ISME J. 2011, 5, 1038–1047. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, W.; Ding, W.; Liang, Z.; Long, L.; Wong, W.C.; Qian, P.-Y. Profiling signal transduction in global marine biofilms. Front. Microbiol. 2022, 12, 768926. [Google Scholar] [CrossRef]
- Trevathan-Tackett, S.M.; Sherman, C.D.; Huggett, M.J.; Campbell, A.H.; Laverock, B.; Hurtado-McCormick, V.; Seymour, J.R.; Firl, A.; Messer, L.F.; Ainsworth, T.D. A horizon scan of priorities for coastal marine microbiome research. Nat. Ecol. Evol. 2019, 3, 1509–1520. [Google Scholar] [CrossRef]
- Hosie, A.H.; Poole, P.S. Bacterial ABC transporters of amino acids. Res. Microbiol. 2001, 152, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, S.E.; Frank, A.M.; Corgliano, D.M.; Seifert, C.; Hauser, L.; Collart, F.R. Environment sensing and response mediated by ABC transporters. BMC Genom. 2011, 12, S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, R.; Sun, J.; Villanueva-Meyer, J.; Mutch, C.; De Los Santos, J.; Peters, J.; Korenchan, D.E.; Neumann, K.; Van Criekinge, M.; Kurhanewicz, J. Detection of bacteria-specific metabolism using hyperpolarized [2-13C] pyruvate. ACS Infect. Dis. 2018, 4, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Fourquez, M.; Devez, A.; Schaumann, A.; Gueneugues, A.; Jouenne, T.; Obernosterer, I.; Blain, S. Effects of iron limitation on growth and carbon metabolism in oceanic and coastal heterotrophic bacteria. Limnol. Oceanogr. 2014, 59, 349–360. [Google Scholar] [CrossRef]
- Hmelo, L.R. Quorum sensing in marine microbial environments. Annu. Rev. Mar. Sci. 2017, 9, 257–281. [Google Scholar] [CrossRef] [PubMed]
- Pachiadaki, M.G.; Sintes, E.; Bergauer, K.; Brown, J.M.; Record, N.R.; Swan, B.K.; Mathyer, M.E.; Hallam, S.J.; Lopez-Garcia, P.; Takaki, Y. Major role of nitrite-oxidizing bacteria in dark ocean carbon fixation. Science 2017, 358, 1046–1051. [Google Scholar] [CrossRef] [Green Version]
- Carter, A.P.; Clemons, W.M.; Brodersen, D.E.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 2000, 407, 340–348. [Google Scholar] [CrossRef]
- Qiao, M.; Ying, G.-G.; Singer, A.C.; Zhu, Y.-G. Review of antibiotic resistance in China and its environment. Environ. Int. 2018, 110, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Singer, A.C.; Shaw, H.; Rhodes, V.; Hart, A. Review of antimicrobial resistance in the environment and its relevance to environmental regulators. Front. Microbiol. 2016, 7, 1728. [Google Scholar] [CrossRef] [Green Version]
- Amarasiri, M.; Sano, D.; Suzuki, S. Understanding human health risks caused by antibiotic resistant bacteria (ARB) and antibiotic resistance genes (ARG) in water environments: Current knowledge and questions to be answered. Crit. Rev. Environ. Sci. Technol. 2020, 50, 2016–2059. [Google Scholar] [CrossRef]
- Larsson, D.J.; Flach, C.-F. Antibiotic resistance in the environment. Nat. Rev. Microbiol. 2022, 20, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Cook, D.N.; Alberti, M.; Pon, N.G.; Nikaido, H.; Hearst, J.E. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol. Microbiol. 1995, 16, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett III, G.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, S.; Behbehani, M.; Habibi, N.; Faizuddin, M.; Al-Murad, M.; Martinez-Guijarro, K.; Al-Sarawi, H.A.; Karam, Q. Microplastics in Kuwait’s Wastewater Streams. Sustainability 2022, 14, 15817. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Observed | Chao 1 | ACE | Shannon | Simpson | Fisher |
|---|---|---|---|---|---|---|
| S1 | 3923 | 4269 | 4272 | 3.94 | 0.776 | 1352 |
| S2 | 4645 | 4953 | 4888 | 4.07 | 0.782 | 1647 |
| S3 | 4774 | 5051 | 5036 | 4.05 | 0.787 | 1609 |
| S4 | 4457 | 4707 | 4711 | 3.83 | 0.772 | 1470 |
| S5 | 4677 | 4907 | 4887 | 3.85 | 0.764 | 1598 |
| S6 | 4785 | 5065 | 5026 | 3.84 | 0.752 | 1731 |
| S7 | 4721 | 5006 | 4990 | 3.90 | 0.768 | 1576 |
| S8 | 4763 | 5038 | 4979 | 4.01 | 0.796 | 1590 |
| S9 | 4478 | 4828 | 4717 | 4.11 | 0.789 | 1433 |
| S10 | 4613 | 4880 | 4857 | 3.94 | 0.781 | 1493 |
| S11 | 4394 | 4645 | 4590 | 3.96 | 0.788 | 1351 |
| S12 | 4452 | 4696 | 4666 | 3.86 | 0.772 | 1440 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habibi, N.; Uddin, S.; Al-Sarawi, H.; Aldhameer, A.; Shajan, A.; Zakir, F.; Abdul Razzack, N.; Alam, F. Metagenomes from Coastal Sediments of Kuwait: Insights into the Microbiome, Metabolic Functions and Resistome. Microorganisms 2023, 11, 531. https://doi.org/10.3390/microorganisms11020531
Habibi N, Uddin S, Al-Sarawi H, Aldhameer A, Shajan A, Zakir F, Abdul Razzack N, Alam F. Metagenomes from Coastal Sediments of Kuwait: Insights into the Microbiome, Metabolic Functions and Resistome. Microorganisms. 2023; 11(2):531. https://doi.org/10.3390/microorganisms11020531
Chicago/Turabian StyleHabibi, Nazima, Saif Uddin, Hanan Al-Sarawi, Ahmed Aldhameer, Anisha Shajan, Farhana Zakir, Nasreem Abdul Razzack, and Faiz Alam. 2023. "Metagenomes from Coastal Sediments of Kuwait: Insights into the Microbiome, Metabolic Functions and Resistome" Microorganisms 11, no. 2: 531. https://doi.org/10.3390/microorganisms11020531