

Characterization of the Gut Microbiota in Urban Thai Individuals Reveals Enterotype-Specific Signature
 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Cohort
2.2. DNA Extraction, Library Preparation, and 16S rDNA Gene Sequencing
2.3. Microbiome Data Analysis
2.4. Enterotyping
2.5. Statistical Analysis
2.6. Microbial Diversity Analysis
2.7. Differential Abundance Analysis
2.8. Co-Occurrence Network Construction and Network Property Measurement
2.9. Microbial Function Prediction and Pathway Enrichment Analysis
3. Results
3.1. Demographic Data of Enrolled Participants from the MODGUT Project
3.2. Thai Gut Microbiota Profiling
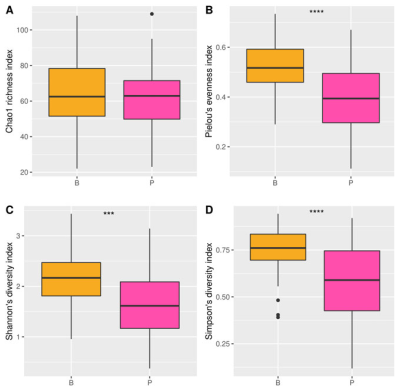
3.3. Association of Microbes and Diversity in Urban Thai Subjects
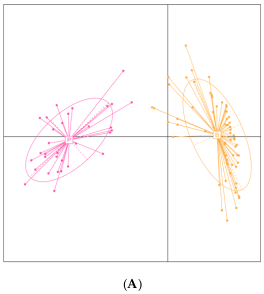
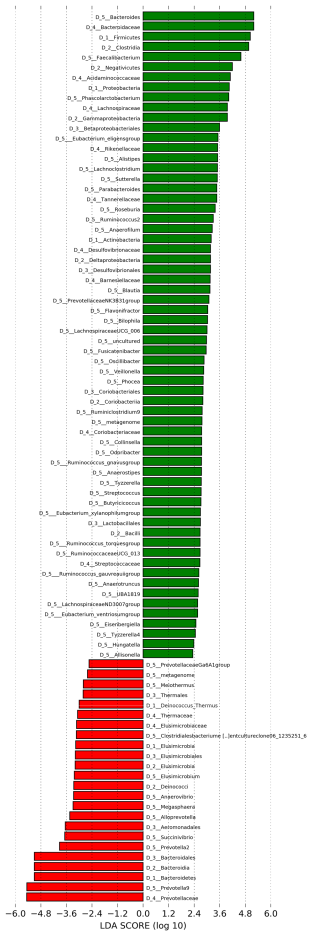
3.4. Stratification and Characterization of Thai Individuals Based on Their Gut Microbiota Profiles
3.5. Microbial Co-Occurrence Network and Clusters of Strongly Related Microbes
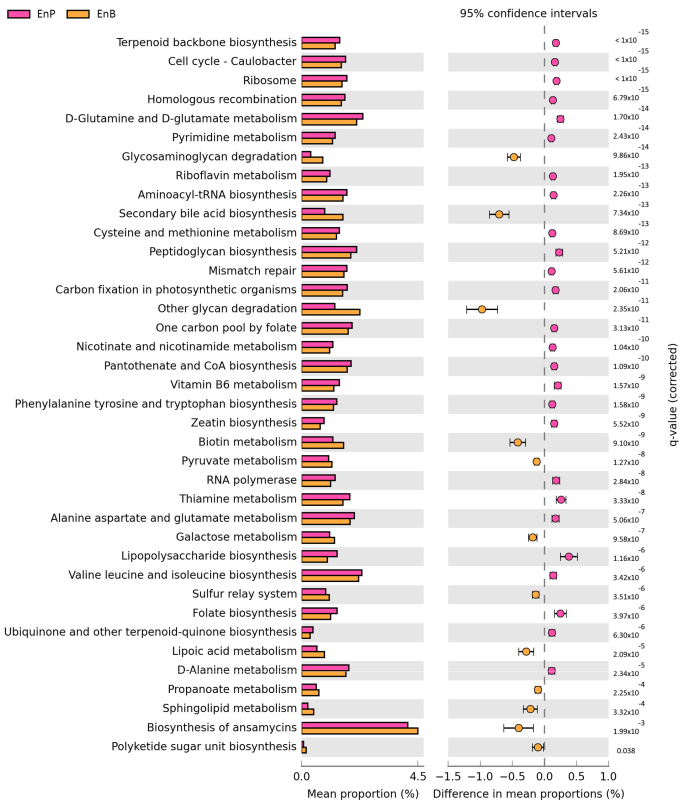
3.6. Metabolic Potential Prediction of Prevotella and Bacteroides Enterotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [Green Version]
- Ridaura, V.; Belkaid, Y. Gut microbiota: The link to your second brain. Cell 2015, 161, 193–194. [Google Scholar] [CrossRef] [Green Version]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Kampmann, C.; Dicksved, J.; Engstrand, L.; Rautelin, H. Composition of human faecal microbiota in resistance to Campylobacter infection. Clin. Microbiol. Infect. 2016, 22, 61.e1–61.e8. [Google Scholar] [CrossRef] [Green Version]
- Gaulke, C.A.; Sharpton, T.J. The influence of ethnicity and geography on human gut microbiome composition. Nat. Med. 2018, 24, 1495–1496. [Google Scholar] [CrossRef]
- Arrieta, M.C.; Stiemsma, L.T.; Amenyogbe, N.; Brown, E.M.; Finlay, B. The intestinal microbiome in early life: Health and disease. Front. Immunol. 2014, 5, 427. [Google Scholar] [CrossRef] [Green Version]
- Salminen, S.; Gibson, G.R.; McCartney, A.L.; Isolauri, E. Influence of mode of delivery on gut microbiota composition in seven year old children. Gut 2004, 53, 1388–1389. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050. [Google Scholar] [CrossRef]
- Wen, L.; Duffy, A. Factors Influencing the Gut Microbiota, Inflammation, and Type 2 Diabetes. J. Nutr. 2017, 147, 1468S–1475S. [Google Scholar] [CrossRef] [Green Version]
- Guarner, F.; Malagelada, J.R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Fan, B.; Liu, W.; Ren, R.; Chen, H.; Bai, S.; Zhu, L.; Sun, G.; Yang, Y.; Wang, X. Investigation into the stability and culturability of Chinese enterotypes. Sci. Rep. 2017, 7, 7947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roager, H.M.; Licht, T.R.; Poulsen, S.K.; Larsen, T.M.; Bahl, M.I. Microbial Enterotypes, Inferred by the Prevotella-to-Bacteroides Ratio, Remained Stable during a 6-Month Randomized Controlled Diet Intervention with the New Nordic Diet. Appl. Environ. Microbiol. 2014, 80, 1142–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, Z.; Lim, A.A.Q.; Zheng, Y.; Koh, E.Y.; Ho, D.; Qiao, J.; Huo, D.; Hou, Q.; Huang, W.; et al. Mongolians core gut microbiota and its correlation with seasonal dietary changes. Sci. Rep. 2014, 4, 5001. [Google Scholar] [CrossRef] [Green Version]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Dhakan, D.B.; Maji, A.; Sharma, A.K.; Saxena, R.; Pulikkan, J.; Grace, T.; Gomez, A.; Scaria, J.; Amato, K.R.; Sharma, V.K. The unique composition of Indian gut microbiome, gene catalogue, and associated fecal metabolome deciphered using multi-omics approaches. Gigascience 2019, 8, giz004. [Google Scholar] [CrossRef]
- Liang, C.; Tseng, H.C.; Chen, H.M.; Wang, W.C.; Chiu, C.M.; Chang, J.Y.; Lu, K.Y.; Weng, S.L.; Chang, T.H.; Chang, C.H.; et al. Diversity and enterotype in gut bacterial community of adults in Taiwan. BMC Genom. 2017, 18, 932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Moraes, A.C.; Fernandes, G.R.; da Silva, I.T.; Almeida-Pititto, B.; Gomes, E.P.; Pereira, A.D.C.; Ferreira, S.R. Enterotype may drive the dietary-associated cardiometabolic risk factors. Front. Cell. Infect. Microbiol. 2017, 7, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Unno, T.; Kang, S.; Park, S. A Korean-Style Balanced Diet Has a Potential Connection with Ruminococcaceae Enterotype and Reduction of Metabolic Syndrome Incidence in Korean Adults. Nutrients 2021, 13, 495. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [Green Version]
- Wutthi-in, M.; Cheevadhanarak, S.; Yasom, S.; Kerdphoo, S.; Thiennimitr, P.; Phrommintikul, A.; Chattipakorn, N.; Kittichotirat, W.; Chattipakorn, S. Gut Microbiota Profiles of Treated Metabolic Syndrome Patients and their Relationship with Metabolic Health. Sci. Rep. 2020, 10, 10085. [Google Scholar] [CrossRef] [PubMed]
- Huse, S.M.; Ye, Y.; Zhou, Y.; Fodor, A.A. A Core Human Microbiome as Viewed through 16S rRNA Sequence Clusters. PLoS ONE 2012, 7, e34242. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Yang, T.W.; Lee, W.H.; Tu, S.J.; Huang, W.C.; Chen, H.M.; Sun, T.H.; Tsai, M.C.; Wang, C.C.; Chen, H.Y.; Huang, C.C.; et al. Enterotype-based Analysis of Gut Microbiota along the Conventional Adenoma-Carcinoma Colorectal Cancer Pathway. Sci. Rep. 2019, 9, 10923. [Google Scholar] [CrossRef] [Green Version]
- Hosoda, S.; Nishijima, S.; Fukunaga, T.; Hattori, M.; Hamada, M. Revealing the microbial assemblage structure in the human gut microbiome using latent Dirichlet allocation. Microbiome 2020, 8, 95. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Zhong, H.; Penders, J.; Shi, Z.; Ren, H.; Cai, K.; Fang, C.; Ding, Q.; Thijs, C.; Blaak, E.E.; Stehouwer, C.D.A.; et al. Impact of early events and lifestyle on the gut microbiota and metabolic phenotypes in young school-age children. Microbiome 2019, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisuse, J.; La-ongkham, O.; Nakphaichit, M.; Therdtatha, P.; Momoda, R.; Tanaka, M.; Fukuda, S.; Popluechai, S.; Kespechara, K.; Sonomoto, K.; et al. Urban Diets Linked to Gut Microbiome and Metabolome Alterations in Children: A Comparative Cross-Sectional Study in Thailand. Front. Microbiol. 2018, 9, 1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Bäckhed, F.; Blaser, M.J.; Bushman, F.D.; de Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovatcheva-Datchary, P.; Nilsson, A.; Akrami, R.; Lee, Y.S.; De Vadder, F.; Arora, T.; Hallen, A.; Martens, E.; Björck, I.; Bäckhed, F. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab. 2015, 22, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Simpson, H.L.; Campbell, B.J. Review article: Dietary fibre–microbiota interactions. Aliment. Pharmacol. Ther. 2015, 42, 158–179. [Google Scholar] [CrossRef] [Green Version]
- Tomova, A.; Bukovsky, I.; Rembert, E.; Yonas, W.; Alwarith, J.; Barnard, N.D.; Kahleova, H. The effects of vegetarian and vegan diets on gut microbiota. Front. Nutr. 2019, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Klimenko, N.S.; Tyakht, A.V.; Popenko, A.S.; Vasiliev, A.S.; Altukhov, I.A.; Ischenko, D.S.; Shashkova, T.I.; Efimova, D.A.; Nikogosov, D.A.; Osipenko, D.A. Microbiome responses to an uncontrolled short-term diet intervention in the frame of the citizen science project. Nutrients 2018, 10, 576. [Google Scholar] [CrossRef] [Green Version]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.K.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef] [Green Version]
- Hyman, R.W.; Fukushima, M.; Jiang, H.; Fung, E.; Rand, L.; Johnson, B.; Vo, K.C.; Caughey, A.B.; Hilton, J.F.; Davis, R.W. Diversity of the vaginal microbiome correlates with preterm birth. Reprod. Sci. 2014, 21, 32–40. [Google Scholar] [CrossRef]
- Brooks, A.W.; Priya, S.; Blekhman, R.; Bordenstein, S.R. Gut microbiota diversity across ethnicities in the United States. PLoS Biol. 2018, 16, e2006842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halton, T.L.; Willett, W.C.; Liu, S.; Manson, J.E.; Stampfer, M.J.; Hu, F.B. Potato and french fry consumption and risk of type 2 diabetes in women. Am. J. Clin. Nutr. 2006, 83, 284–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.N.; Roy, S.K.; Rahaman, M.A. Epidemiological predictors of metabolic syndrome in urban West Bengal, India. J. Fam. Med. Prim. Care 2015, 4, 535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omuse, G.; Maina, D.; Hoffman, M.; Mwangi, J.; Wambua, C.; Kagotho, E.; Amayo, A.; Ojwang, P.; Premji, Z.; Ichihara, K. Metabolic syndrome and its predictors in an urban population in Kenya: A cross sectional study. BMC Endocr. Disord. 2017, 17, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundarakumar, J.S.; Stezin, A.; Menesgere, A.L.; Ravindranath, V.; SANSCOG; TLSA Collaborators. Rural-urban and gender differences in metabolic syndrome in the aging population from southern India: Two parallel, prospective cohort studies. EClinicalMedicine 2022, 47, 101395. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 July 2010).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2013, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Caliński, T.; Harabasz, J. A dendrite method for cluster analysis. Commun. Stat.-Theory Methods 1974, 3, 1–27. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Endres, D.M.; Schindelin, J.E. A new metric for probability distributions. IEEE Trans. Inf. Theory 2003, 49, 1858–1860. [Google Scholar] [CrossRef] [Green Version]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience 2013, 2, 2047-217X-2-16. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Akoglu, H. User’s guide to correlation coefficients. Turk. J. Emerg. Med. 2018, 18, 91–93. [Google Scholar] [CrossRef]
- Li, M.; Li, D.; Tang, Y.; Wu, F.; Wang, J. CytoCluster: A cytoscape plugin for cluster analysis and visualization of biological networks. Int. J. Mol. Sci. 2017, 18, 1880. [Google Scholar] [CrossRef] [Green Version]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Baralis, E.; Bianco, A.; Cerquitelli, T.; Chiaraviglio, L.; Mellia, M. NetCluster: A Clustering-Based Framework for Internet Tomography. In Proceedings of the 2009 IEEE International Conference on Communications, Dresden, Germany, 14–18 June 2009; pp. 1–5. [Google Scholar]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.J.; Berger, J.O.; Johannesson, M.; Nosek, B.A.; Wagenmakers, E.J.; Berk, R.; Bollen, K.A.; Brembs, B.; Brown, L.; Camerer, C.; et al. Redefine statistical significance. Nat. Hum. Behav. 2018, 2, 6–10. [Google Scholar] [CrossRef] [Green Version]
- García-López, R.; Cornejo-Granados, F.; Lopez-Zavala, A.A.; Sánchez-López, F.; Cota-Huízar, A.; Sotelo-Mundo, R.R.; Guerrero, A.; Mendoza-Vargas, A.; Gómez-Gil, B.; Ochoa-Leyva, A. Doing More with Less: A Comparison of 16S Hypervariable Regions in Search of Defining the Shrimp Microbiota. Microorganisms 2020, 8, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, D.; Yang, L.; Pei, Z. Gut Microbiota, Fusobacteria, and Colorectal Cancer. Diseases 2018, 6, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broadley, M.; Schweon, S.J. Get the facts about Fusobacterium. Nursing2020 2017, 47, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef]
- Asgari, Y.; Salehzadeh-Yazdi, A.; Schreiber, F.; Masoudi-Nejad, A. Controllability in cancer metabolic networks according to drug targets as driver nodes. PLoS ONE 2013, 8, e79397. [Google Scholar] [CrossRef] [Green Version]
- Pavlopoulos, G.A.; Secrier, M.; Moschopoulos, C.N.; Soldatos, T.G.; Kossida, S.; Aerts, J.; Schneider, R.; Bagos, P.G. Using graph theory to analyze biological networks. BioData Min. 2011, 4, 10. [Google Scholar] [CrossRef]
- Agrawal, S.; Agrawal, A.; Said, H.M. Biotin deficiency enhances the inflammatory response of human dendritic cells. Am. J. Physiol.-Cell Physiol. 2016, 311, C386–C391. [Google Scholar] [CrossRef] [Green Version]
- Hjorth, M.F.; Blædel, T.; Bendtsen, L.Q.; Lorenzen, J.K.; Holm, J.B.; Kiilerich, P.; Roager, H.M.; Kristiansen, K.; Larsen, L.H.; Astrup, A. Prevotella-to-Bacteroides ratio predicts body weight and fat loss success on 24-week diets varying in macronutrient composition and dietary fiber: Results from a post-hoc analysis. Int. J. Obes. 2019, 43, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Sianoya, A.; Mishima, R.; Therdtatha, P.; Rodriguez, A.; Ramos, D.C.; Lee, Y.K.; Dalmacio, L.M.; Nakayama, J. Gut microbiome status of urban and rural Filipino adults in relation to diet and metabolic disorders. FEMS Microbiol. Lett. 2021, 368, fnab149. [Google Scholar] [CrossRef]
- Rosas-Plaza, S.; Hernández-Terán, A.; Navarro-Díaz, M.; Escalante, A.E.; Morales-Espinosa, R.; Cerritos, R. Human Gut Microbiome Across Different Lifestyles: From Hunter-Gatherers to Urban Populations. Front. Microbiol. 2022, 13, 843170. [Google Scholar] [CrossRef]
- Hjorth, M.; Roager, H.M.; Larsen, T.; Poulsen, S.; Licht, T.R.; Bahl, M.I.; Zohar, Y.; Astrup, A. Pre-treatment microbial Prevotella-to-Bacteroides ratio, determines body fat loss success during a 6-month randomized controlled diet intervention. Int. J. Obes. 2018, 42, 580–583. [Google Scholar] [CrossRef] [Green Version]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiumento, S.; Roblin, C.; Kieffer-Jaquinod, S.; Tachon, S.; Leprètre, C.; Basset, C.; Aditiyarini, D.; Olleik, H.; Nicoletti, C.; Bornet, O.; et al. Ruminococcin C, a promising antibiotic produced by a human gut symbiont. Sci. Adv. 2019, 5, eaaw9969. [Google Scholar] [CrossRef] [Green Version]
- Ze, X.; Duncan, S.H.; Louis, P.; Flint, H.J. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012, 6, 1535–1543. [Google Scholar] [CrossRef] [Green Version]
- La Reau, A.J.; Suen, G. The Ruminococci: Key symbionts of the gut ecosystem. J. Microbiol. 2018, 56, 199–208. [Google Scholar] [CrossRef]
- Scott, K.P.; Jean-Michel, A.; Midtvedt, T.; van Hemert, S. Manipulating the gut microbiota to maintain health and treat disease. Microb. Ecol. Health Dis. 2015, 26, 25877. [Google Scholar] [CrossRef]
- Meslier, V.; Laiola, M.; Roager, H.M.; De Filippis, F.; Roume, H.; Quinquis, B.; Giacco, R.; Mennella, I.; Ferracane, R.; Pons, N.; et al. Mediterranean diet intervention in overweight and obese subjects lowers plasma cholesterol and causes changes in the gut microbiome and metabolome independently of energy intake. Gut 2020, 69, 1258–1268. [Google Scholar] [CrossRef] [Green Version]
- Hagerty, S.L.; Hutchison, K.E.; Lowry, C.A.; Bryan, A.D. An empirically derived method for measuring human gut microbiome alpha diversity: Demonstrated utility in predicting health-related outcomes among a human clinical sample. PLoS ONE 2020, 15, e0229204. [Google Scholar] [CrossRef] [Green Version]
- Menni, C.; Jackson, M.A.; Pallister, T.; Steves, C.J.; Spector, T.D.; Valdes, A.M. Gut microbiome diversity and high-fibre intake are related to lower long-term weight gain. Int. J. Obes. 2017, 41, 1099–1105. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Yang, W.; Chen, G.; Shafiq, M.; Javed, S.; Zaidi, S.S.A.; Shahid, R.; Liu, C.; Bokhari, H. Analysis of gut microbiota of obese individuals with type 2 diabetes and healthy individuals. PLoS ONE 2019, 14, e0226372. [Google Scholar] [CrossRef] [Green Version]
- Almugadam, B.S.; Liu, Y.; Chen, S.M.; Wang, C.H.; Shao, C.Y.; Ren, B.W.; Tang, L. Alterations of Gut Microbiota in Type 2 Diabetes Individuals and the Confounding Effect of Antidiabetic Agents. J. Diabetes Res. 2020, 2020, 7253978. [Google Scholar] [CrossRef]
- Afra, K.; Laupland, K.; Leal, J.; Lloyd, T.; Gregson, D. Incidence, risk factors, and outcomes of Fusobacterium species bacteremia. BMC Infect Dis. 2013, 13, 264. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.J.; Li, S.; Gan, R.Y.; Zhou, T.; Xu, D.P.; Li, H.B. Impacts of Gut Bacteria on Human Health and Diseases. Int. J. Mol. Sci. 2015, 16, 7493–7519. [Google Scholar] [CrossRef]
- Wan, L.; Ge, W.R.; Zhang, S.; Sun, Y.L.; Wang, B.; Yang, G. Case-Control Study of the Effects of Gut Microbiota Composition on Neurotransmitter Metabolic Pathways in Children With Attention Deficit Hyperactivity Disorder. Front. Neurosci. 2020, 14, 127. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Hart, R.A.; Charlesworth, R.P.G.; Sharpley, C.F. Gut microbiome and depression: What we know and what we need to know. Rev. Neurosci. 2018, 29, 629–643. [Google Scholar] [CrossRef]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.D.; Peng, X.B.; Zhao, R.R.; Ma, C.Q.; Li, J.N.; Yao, L.Q. The intestinal microbial community dissimilarity in hepatitis B virus-related liver cirrhosis patients with and without at alcohol consumption. Gut Pathog. 2019, 11, 58. [Google Scholar] [CrossRef]
- Kelly, T.N.; Bazzano, L.A.; Ajami, N.J.; He, H.; Zhao, J.; Petrosino, J.F.; Correa, A.; He, J. Gut Microbiome Associates With Lifetime Cardiovascular Disease Risk Profile Among Bogalusa Heart Study Participants. Circ. Res. 2016, 119, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, Q.; Lin, P.; Xu, R.; He, D.; Ji, W.; Bian, Y.; Shen, Y.; Li, Q.; Liu, C.; et al. Characteristics of Gut Microbiota in Patients With Rheumatoid Arthritis in Shanghai, China. Front. Cell. Infect. Microbiol. 2019, 9, 369. [Google Scholar] [CrossRef] [Green Version]
- Signat, B.; Roques, C.; Poulet, P.; Duffaut, D. Fusobacterium nucleatum in Periodontal Health and Disease. Curr. Issues Mol. Biol. 2011, 13, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Amitay, E.L.; Werner, S.; Vital, M.; Pieper, D.H.; Höfler, D.; Gierse, I.J.; Butt, J.; Balavarca, Y.; Cuk, K.; Brenner, H. Fusobacterium and colorectal cancer: Causal factor or passenger? Results from a large colorectal cancer screening study. Carcinogenesis 2017, 38, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Wang, R.; Bhattacharya, R.; Boulbes, D.R.; Fan, F.; Xia, L.; Adoni, H.; Ajami, N.J.; Wong, M.C.; Smith, D.P.; et al. Fusobacterium Nucleatum Subspecies Animalis Influences Proinflammatory Cytokine Expression and Monocyte Activation in Human Colorectal Tumors. Cancer Prev. Res. 2017, 10, 398–409. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Yang, X.; Zhang, R.; Li, J.; Xiao, X.; Hu, Y.; Chen, Y.; Yang, F.; Lu, N.; Wang, Z.; et al. Dysbiosis Signature of Fecal Microbiota in Colorectal Cancer Patients. Microb. Ecol. 2013, 66, 462–470. [Google Scholar] [CrossRef]
- Manson McGuire, A.; Cochrane, K.; Griggs, A.D.; Haas, B.J.; Abeel, T.; Zeng, Q.; Nice, J.B.; MacDonald, H.; Birren, B.W.; Berger, B.W.; et al. Evolution of invasion in a diverse set of Fusobacterium species. MBio 2014, 5, e01864. [Google Scholar] [CrossRef] [Green Version]
- Magnúsdóttir, S.; Ravcheev, D.; de Crécy-Lagard, V.; Thiele, I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front. Genet. 2015, 6, 148. [Google Scholar] [CrossRef]
- Rothstein, D.M. Rifamycins, Alone and in Combination. Cold Spring Harb. Perspect. Med. 2016, 6, a027011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Oh, J.H.; Chung, J.H. Glycosaminoglycan and proteoglycan in skin aging. J. Dermatol. Sci. 2016, 83, 174–181. [Google Scholar] [CrossRef]
- Zeng, H.; Umar, S.; Rust, B.; Lazarova, D.; Bordonaro, M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int. J. Mol. Sci. 2019, 20, 1214. [Google Scholar] [CrossRef] [Green Version]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.F.; Cravetto, C.; Molino, G.; Belforte, G.; Bona, B. Simulation of the metabolism and enterohepatic circulation of endogenous deoxycholic acid in humans using a physiologic pharmacokinetic model for bile acid metabolism. Gastroenterology 1987, 93, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.; Holubec, H.; Bhattacharyya, A.K.; Nguyen, H.; Payne, C.M.; Zaitlin, B.; Bernstein, H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch. Toxicol. 2011, 85, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.R.; Haileselassie, Y.; Nguyen, L.P.; Tropini, C.; Wang, M.; Becker, L.S.; Sim, D.; Jarr, K.; Spear, E.T.; Singh, G. Dysbiosis-induced secondary bile acid deficiency promotes intestinal inflammation. Cell Host Microbe 2020, 27, 659–670. [Google Scholar] [CrossRef]
- Milovic, V.; Teller, I.C.; Faust, D.; Caspary, W.F.; Stein, J. Effects of deoxycholate on human colon cancer cells: Apoptosis or proliferation. Eur. J. Clin. Investig. 2002, 32, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.T.; Thiele, I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 2019, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Naqvi, A.; Rangwala, H.; Keshavarzian, A.; Gillevet, P. Network-Based Modeling of the Human Gut Microbiome Chem. Biodivers. 2010, 7, 1040–1050. [Google Scholar] [CrossRef]
- Wexler, A.G.; Goodman, A.L. An insider’s perspective: Bacteroides as a window into the microbiome. Nat. Microbiol. 2017, 2, 17026. [Google Scholar] [CrossRef] [Green Version]
- Wexler, H.M. Bacteroides: The Good, the Bad, and the Nitty-Gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djekic, D.; Shi, L.; Brolin, H.; Carlsson, F.; Särnqvist, C.; Savolainen, O.; Cao, Y.; Bäckhed, F.; Tremaroli, V.; Landberg, R. Effects of a vegetarian diet on cardiometabolic risk factors, gut microbiota, and plasma metabolome in subjects with ischemic heart disease: A randomized, crossover study. J. Am. Heart Assoc. 2020, 9, e016518. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Hosomi, K.; Kawashima, H.; Chen, Y.A.; Mohsen, A.; Ohno, H.; Konishi, K.; Tanisawa, K.; Kifushi, M.; Kogawa, M. Dietary Vitamin B1 Intake Influences Gut Microbial Community and the Consequent Production of Short-Chain Fatty Acids. Nutrients 2022, 14, 2078. [Google Scholar] [CrossRef]
- van Trijp, M.P.H.; Schutte, S.; Esser, D.; Wopereis, S.; Hoevenaars, F.P.M.; Hooiveld, G.J.E.J.; Afman, L.A. Minor Changes in the Composition and Function of the Gut Microbiota During a 12-Week Whole Grain Wheat or Refined Wheat Intervention Correlate with Liver Fat in Overweight and Obese Adults. J. Nutr. 2021, 151, 491–502. [Google Scholar] [CrossRef]
- Kosulwat, V. The nutrition and health transition in Thailand. Public Health Nutr. 2002, 5, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Nagao-Kitamoto, H.; Kamada, N. Host-microbial Cross-talk in Inflammatory Bowel Disease. Immune Netw. 2017, 17, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.X.; Thomas, C.E.; Brunak, S. Network biology concepts in complex disease comorbidities. Nat. Rev. Genet. 2016, 17, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Landi, P.; Minoarivelo, H.O.; Brännström, Å.; Hui, C.; Dieckmann, U. Complexity and stability of ecological networks: A review of the theory. Popul. Ecol. 2018, 60, 319–345. [Google Scholar] [CrossRef]
- Aliyu, S.H.; Marriott, R.K.; Curran, M.D.; Parmar, S.; Bentley, N.; Brown, N.M.; Brazier, J.S.; Ludlam, H. Real-time PCR investigation into the importance of Fusobacterium necrophorum as a cause of acute pharyngitis in general practice. J. Med. Microbiol. 2004, 53, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, K.Q.; Almeida-da-Silva, C.L.C.; Coutinho-Silva, R. Immunological pathways triggered by Porphyromonas gingivalis and Fusobacterium nucleatum: Therapeutic possibilities? Mediat. Inflamm. 2019, 2019, 7241312. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lifestyle Variables | N (n = 96) | EnP (n = 32) | EnB (n = 64) | p-Value 1 |
|---|---|---|---|---|
| Gender | 0.339 | |||
| Male | 29 | 12 | 17 | |
| Female | 61 | 17 | 44 | |
| Age (years) | 34.17 ± 10.57 | 34.31 ± 10.75 | 34.10 ± 10.56 | 0.972 |
| 18 to 25 | 20 | 6 | 14 | |
| 26 to 35 | 29 | 10 | 19 | |
| 36 to 45 | 31 | 10 | 21 | |
| 46 to 55 | 6 | 2 | 4 | |
| Over 55 | 4 | 1 | 3 | |
| BMI 2 (kg/m2) | 22.58 ± 3.68 | 22.78 ± 3.40 | 22.48 ± 3.82 | 0.884 |
| Underweight | 9 | 2 | 7 | |
| Normal | 48 | 16 | 32 | |
| Overweight | 23 | 8 | 15 | |
| Obese | 10 | 3 | 7 | |
| Types of diets | 0.586 | |||
| Vegetable and animal meat | 62 | 22 | 40 | |
| Vegetable and animal meat but not consume raw meat | 27 | 7 | 20 | |
| Vegan | 1 | 0 | 1 |
| Network Properties | EnP | EnB |
|---|---|---|
| Number of nodes | 67 | 42 |
| Number of edges | 181 (Positive: 166, Negative: 15) | 109 (Positive: 100, Negative: 9) |
| Clustering coefficient | 0.353 | 0.485 |
| Network diameter | 7 | 5 |
| Average number of neighbors | 6.069 | 6.516 |
| Network density | 0.106 | 0.217 |
| Network centralization | 0.271 | 0.409 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinsuebchuea, J.; Paenkaew, P.; Wutthiin, M.; Nantanaranon, T.; Laeman, K.; Kittichotirat, W.; Wattanachaisaereekul, S.; Dulsawat, S.; Nopharatana, M.; Vorapreeda, N.; et al. Characterization of the Gut Microbiota in Urban Thai Individuals Reveals Enterotype-Specific Signature. Microorganisms 2023, 11, 136. https://doi.org/10.3390/microorganisms11010136
Sinsuebchuea J, Paenkaew P, Wutthiin M, Nantanaranon T, Laeman K, Kittichotirat W, Wattanachaisaereekul S, Dulsawat S, Nopharatana M, Vorapreeda N, et al. Characterization of the Gut Microbiota in Urban Thai Individuals Reveals Enterotype-Specific Signature. Microorganisms. 2023; 11(1):136. https://doi.org/10.3390/microorganisms11010136
Chicago/Turabian StyleSinsuebchuea, Jiramaetha, Prasobsook Paenkaew, Montree Wutthiin, Thatchawanon Nantanaranon, Kiattiyot Laeman, Weerayuth Kittichotirat, Songsak Wattanachaisaereekul, Sudarat Dulsawat, Montira Nopharatana, Namol Vorapreeda, and et al. 2023. "Characterization of the Gut Microbiota in Urban Thai Individuals Reveals Enterotype-Specific Signature" Microorganisms 11, no. 1: 136. https://doi.org/10.3390/microorganisms11010136