
Metagenomic Strain-Typing Combined with Isolate Sequencing Provides Increased Resolution of the Genetic Diversity of Campylobacter jejuni Carriage in Wild Birds
 , and
, and
Abstract
:1. Introduction
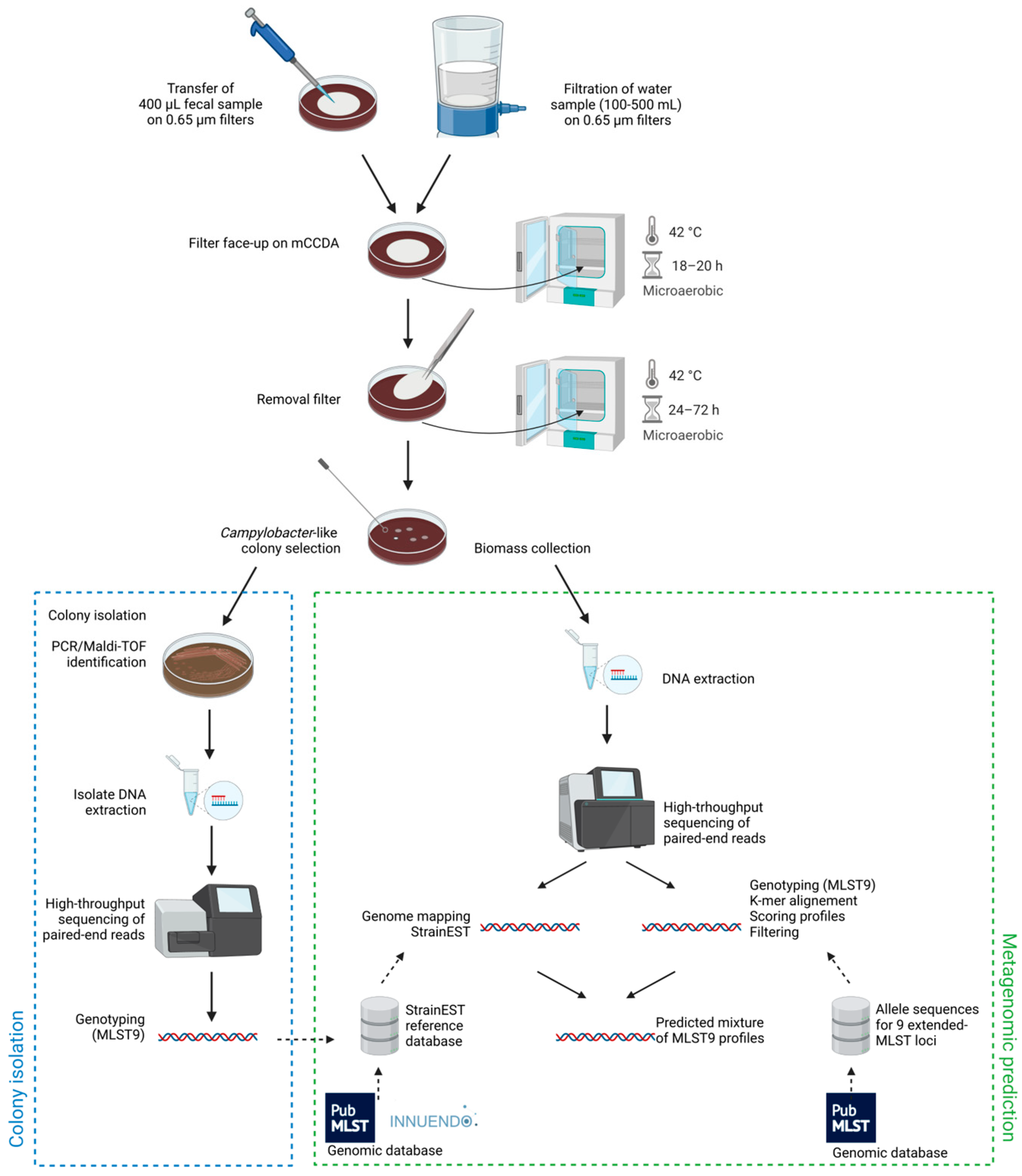
2. Materials and Methods
2.1. Wild Bird Feces and Surface Water Sampling
2.2. Passive Filtration Method (PFM)
2.3. Identification and Sequencing of Campylobacter Isolates
2.4. Sequencing of Filter Sweeps and Validation Samples
2.5. Genotyping of Metagenomic Samples from Filter Sweeps
2.5.1. Preprocessing
2.5.2. K-Mer Alignment (KMA) Step
2.5.3. StrainEST Step
2.5.4. Combining KMA and StrainEST Outputs
2.6. Statistical Analyses and Comparison of Known Mixtures—Recovered Isolates
3. Results
3.1. Overview of Isolated Strains
3.2. Overview of Metagenomic Sequencing Data
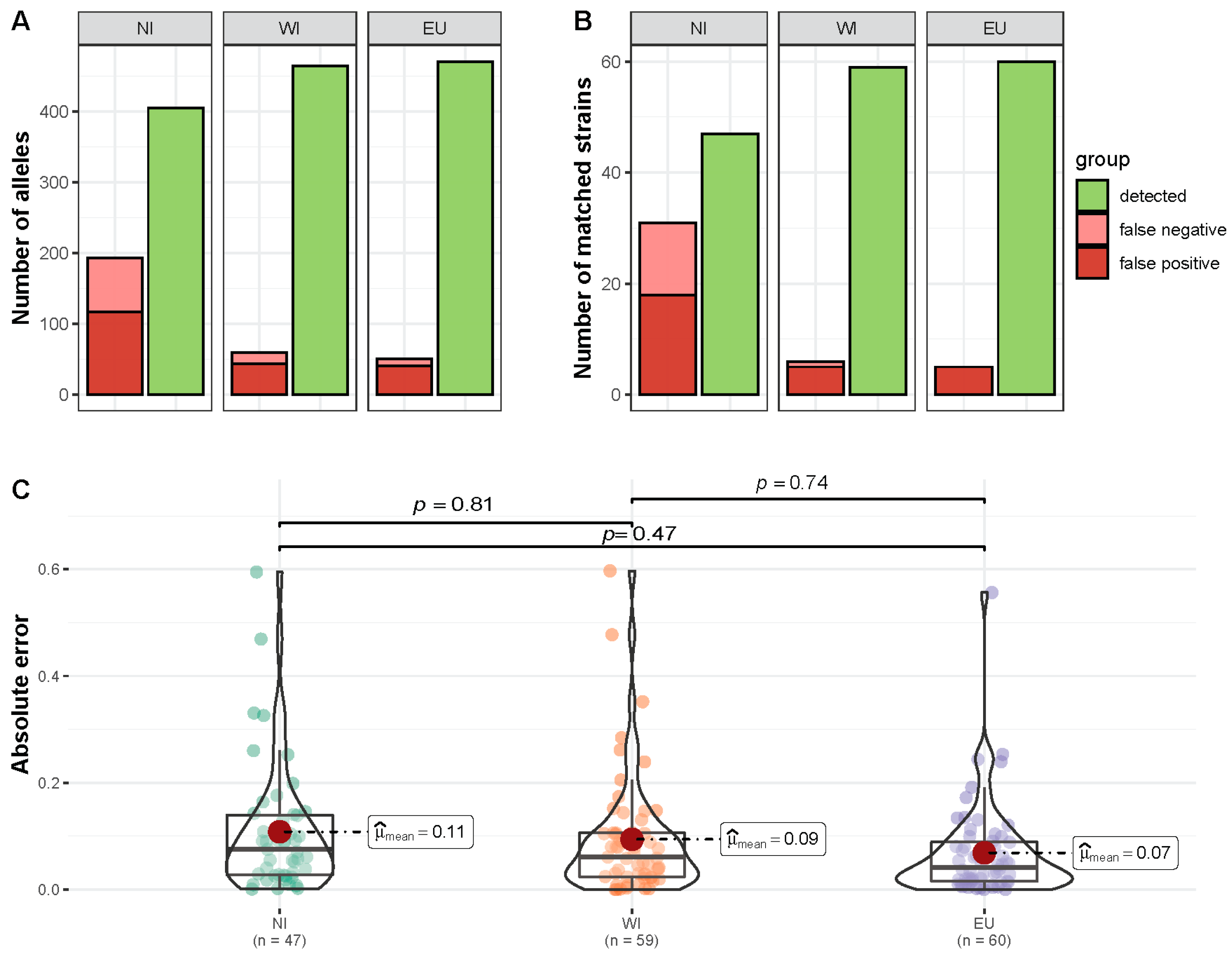
3.3. Prediction of C. jejuni Profiles from Artificial Mixed Samples
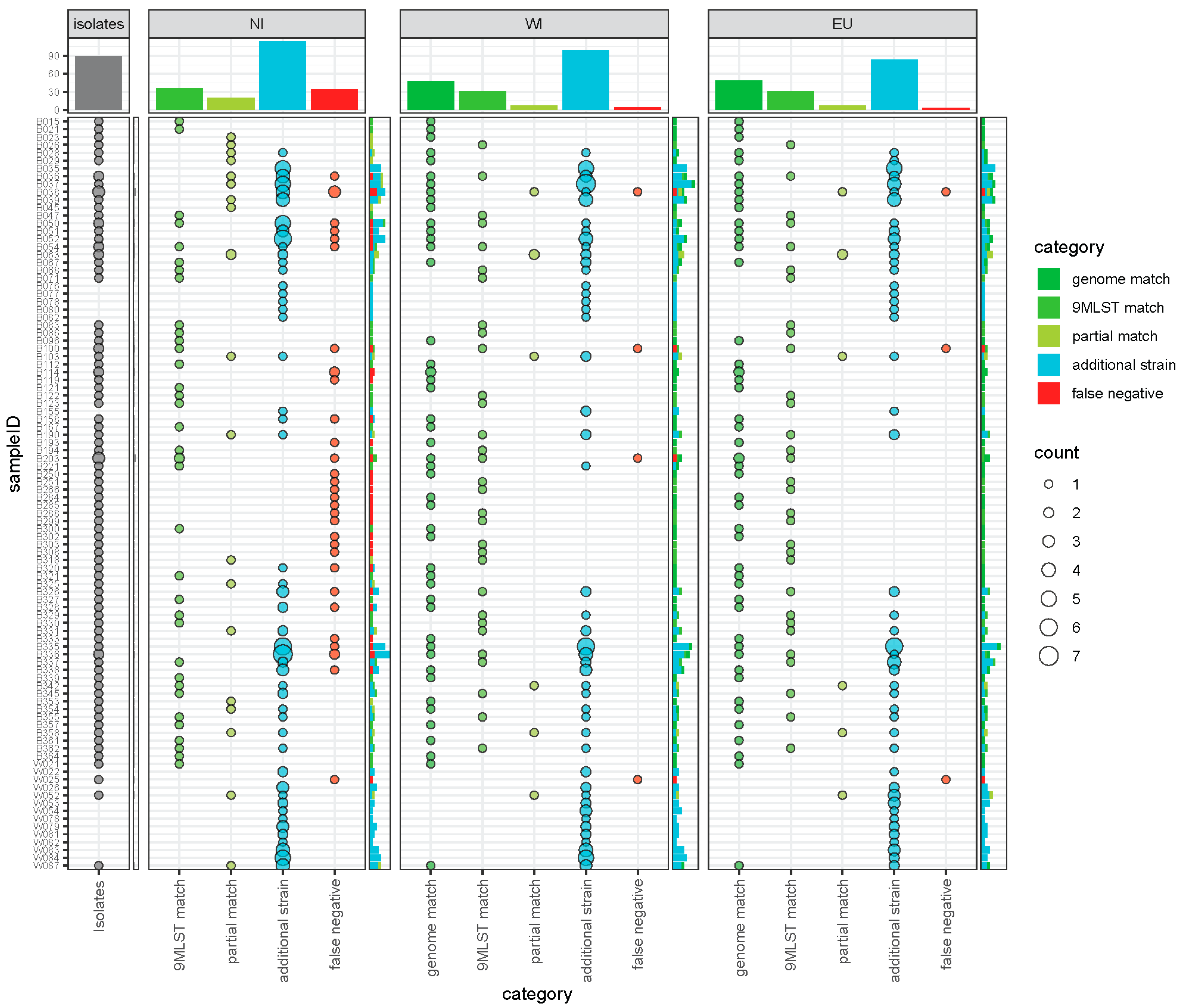
3.4. Comparison of Recovered Isolates and Predicted Profiles
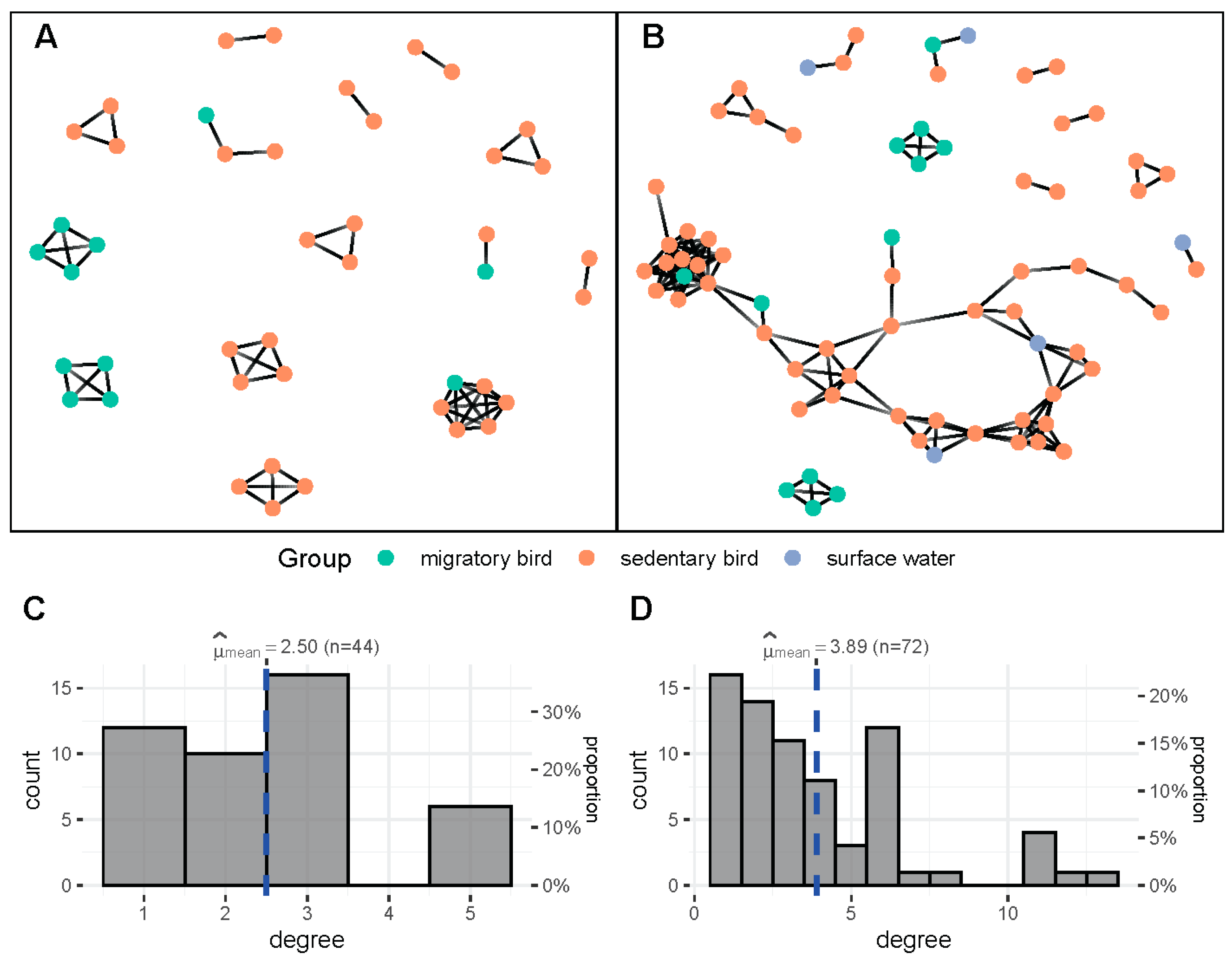
3.5. Assessment of Additional Diversity Detected by Metagenomic Sequencing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Food Safety Authority; European Centre for Disease Prevention and Control. The European Union One Health 2020 Zoonoses Report. EFSA J. 2021, 19, e06971. [Google Scholar] [CrossRef]
- Frosth, S.; Karlsson-Lindsjö, O.; Niazi, A.; Fernström, L.L.; Hansson, I. Identification of Transmission Routes of Campylobacter and On-Farm Measures to Reduce Campylobacter in Chicken. Pathogens 2020, 9, 363. [Google Scholar] [CrossRef] [PubMed]
- Jones, K. Campylobacters in Water, Sewage and the Environment. Symp. Ser. Soc. Appl. Microbiol. 2001, 90, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Vidal, A.B.; Colles, F.M.; Rodgers, J.D.; McCarthy, N.D.; Davies, R.H.; Maiden, M.C.J.; Clifton-Hadley, F.A. Genetic Diversity of Campylobacter jejuni and Campylobacter coli Isolates from Conventional Broiler Flocks and the Impacts of Sampling Strategy and Laboratory Method. Appl. Environ. Microbiol. 2016, 82, 2347–2355. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.A.; Gulhan, T. Campylobacter in Wild Birds: Is It an Animal and Public Health Concern? Front. Microbiol. 2022, 12, 812591. [Google Scholar] [CrossRef] [PubMed]
- Weis, A.M.; Miller, W.A.; Byrne, B.A.; Chouicha, N.; Boyce, W.M.; Townsend, A.K. Prevalence and Pathogenic Potential of Campylobacter Isolates from Free-Living, Human-Commensal American Crows. Appl. Environ. Microbiol. 2014, 80, 1639–1644. [Google Scholar] [CrossRef] [Green Version]
- Mencía-Gutiérrez, A.; Martín-Maldonado, B.; Pastor-Tiburón, N.; Moraleda, V.; González, F.; García-Peña, F.J.; Pérez-Cobo, I.; Revuelta, L.; Marín, M. Prevalence and Antimicrobial Resistance of Campylobacter from Wild Birds of Prey in Spain. Comp. Immunol. Microbiol. Infect. Dis. 2021, 79, 101712. [Google Scholar] [CrossRef]
- ISO 17795; Water Quality—Detection and Enumeration of Thermotolerant Campylobacter Species. International Organization for Standardization: Geneva, Switzerland, 2019.
- ISO 10272-2; Microbiology of the Food Chain-Horizontal Method for Detection and Enumeration of Campylobacter spp. International Organization for Standardization: Geneva, Switzerland, 2017.
- Harrison, L.M.; Balan, K.V.; Hiett, K.L.; Babu, U.S. Current Methodologies and Future Direction of Campylobacter Isolation and Detection from Food Matrices, Clinical Samples, and the Agricultural Environment. J. Microbiol. Methods 2022, 201, 106562. [Google Scholar] [CrossRef]
- Williams, L.K.; Sait, L.C.; Cogan, T.A.; Jørgensen, F.; Grogono-Thomas, R.; Humphrey, T.J. Enrichment Culture Can Bias the Isolation of Campylobacter subtypes. Epidemiol. Infect. 2012, 140, 1227–1235. [Google Scholar] [CrossRef]
- Hetman, B.M.; Mutschall, S.K.; Carrillo, C.D.; Thomas, J.E.; Gannon, V.P.J.; Inglis, G.D.; Taboada, E.N. “These Aren’t the Strains You’re Looking for”: Recovery Bias of Common Campylobacter jejuni Subtypes in Mixed Cultures. Front. Microbiol. 2020, 11, 541. [Google Scholar] [CrossRef]
- Steele, T.W.; McDermott, S.N. The Use of Membrane Filters Applied Directly to the Surface of Agar Plates for the Isolation of Campylobacter jejuni from Feces. Pathology 1984, 16, 263–265. [Google Scholar] [CrossRef] [PubMed]
- On, S.L.W. Isolation, Identification and Subtyping of Campylobacter: Where to from Here? J. Microbiol. Methods 2013, 95, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.G.; Taboada, E.; Grant, C.C.R.; Blakeston, C.; Pollari, F.; Marshall, B.; Rahn, K.; MacKinnon, J.; Daignault, D.; Pillai, D.; et al. Comparison of Molecular Typing Methods Useful for Detecting Clusters of Campylobacter jejuni and C. Coli Isolates through Routine Surveillance. J. Clin. Microbiol. 2012, 50, 798–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, S.K.; Dallas, J.F.; Strachan, N.J.C.; MacRae, M.; McCarthy, N.D.; Wilson, D.J.; Gormley, F.J.; Falush, D.; Ogden, I.D.; Maiden, M.C.J.; et al. Campylobacter Genotyping to Determine the Source of Human Infection. Clin. Infect. Dis. 2009, 48, 1072–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragimbeau, C.; Colin, S.; Devaux, A.; Decruyenaere, F.; Cauchie, H.-M.; Losch, S.; Penny, C.; Mossong, J. Investigating the Host Specificity of Campylobacter jejuni and Campylobacter coli by Sequencing Gyrase Subunit A. BMC Microbiol. 2014, 14, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nennig, M.; Llarena, A.-K.; Herold, M.; Mossong, J.; Penny, C.; Losch, S.; Tresse, O.; Ragimbeau, C. Investigating Major Recurring Campylobacter jejuni Lineages in Luxembourg Using Four Core or Whole Genome Sequencing Typing Schemes. Front. Cell. Infect. Microbiol. 2021, 10, 608020. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Rodriguez-mueller, B.; Garud, N.; Pollard, K.S. An Integrated Metagenomics Pipeline for Strain Profiling Reveals Novel Patterns of Bacterial Transmission and Biogeography. Genome Res. 2016, 26, 1612–1625. [Google Scholar] [CrossRef] [Green Version]
- Quince, C.; Delmont, T.O.; Raguideau, S.; Alneberg, J.; Darling, A.E.; Collins, G.; Eren, A.M. DESMAN: A New Tool for de Novo Extraction of Strains from Metagenomes. Genome Biol. 2017, 18, 181. [Google Scholar] [CrossRef] [Green Version]
- Vicedomini, R.; Quince, C.; Darling, A.E.; Chikhi, R. Strainberry: Automated Strain Separation in Low-Complexity Metagenomes Using Long Reads. Nat. Commun. 2021, 12, 4485. [Google Scholar] [CrossRef]
- Quince, C.; Nurk, S.; Raguideau, S.; James, R.; Soyer, O.S.; Summers, J.K.; Limasset, A.; Eren, A.M.; Chikhi, R.; Darling, A.E. STRONG: Metagenomics Strain Resolution on Assembly Graphs. Genome Biol. 2021, 22, 214. [Google Scholar] [CrossRef]
- Zolfo, M.; Tett, A.; Jousson, O.; Donati, C.; Segata, N. MetaMLST: Multi-Locus Strain-Level Bacterial Typing from Metagenomic Samples. Nucleic Acids Res. 2017, 45, e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, K.; Pons, N.; Berland, M.; Oñate, F.P.; Almeida, M.; Peterlongo, P. StrainFLAIR: Strain-Level Profiling of Metagenomic Samples Using Variation Graphs. PeerJ 2021, 9, e11884. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, S.K.; McCarthy, N.D.; Jolley, K.A.; Maiden, M.C.J. Introgression in the Genus Campylobacter: Generation and Spread of Mosaic Alleles. Microbiology 2011, 157, 1066–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gripp, E.; Hlahla, D.; Didelot, X.; Kops, F.; Maurischat, S.; Tedin, K.; Alter, T.; Ellerbroek, L.; Schreiber, K.; Schomburg, D.; et al. Closely Related Campylobacter jejuni Strains from Different Sources Reveal a Generalist Rather than a Specialist Lifestyle. BMC Genom. 2011, 12, 584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäklin, T.; Kallonen, T.; David, S.; Boinett, C.J.; Pascoe, B.; Méric, G.; Aanensen, D.M.; Feil, E.J.; Baker, S.; Parkhill, J.; et al. High-Resolution Sweep Metagenomics Using Fast Probabilistic Inference. Wellcome Open Res. 2021, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Linton, D.; Lawson, A.J.; Owen, R.J.; Stanley, J. PCR Detection, Identification to Species Level, and Fingerprinting of Campylobacter jejuni and Campylobacter coli Direct from Diarrheic Samples. J. Clin. Microbiol. 1997, 35, 2568–2572. [Google Scholar] [CrossRef] [Green Version]
- Bessède, E.; Solecki, O.; Sifré, E.; Labadi, L.; Mégraud, F. Identification of Campylobacter Species and Related Organisms by Matrix Assisted Laser Desorption Ionization–Time of Flight (MALDI-TOF) Mass Spectrometry. Clin. Microbiol. Infect. 2011, 17, 1735–1739. [Google Scholar] [CrossRef] [Green Version]
- Gorman, R.; Adley, C.C. An Evaluation of Five Preservation Techniques and Conventional Freezing Temperatures of -20oC and -85oC for Long-Term Preservation of Campylobacter jejuni. Lett. Appl. Microbiol. 2004, 38, 306–310. [Google Scholar] [CrossRef]
- Jünemann, S.; Sedlazeck, F.J.; Prior, K.; Albersmeier, A.; John, U.; Kalinowski, J.; Mellmann, A.; Goesmann, A.; von Haeseler, A.; Stoye, J.; et al. Updating Benchtop Sequencing Performance Comparison. Nat. Biotechnol. 2013, 31, 294–296. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 December 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Mölder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-Tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Sustainable Data Analysis with Snakemake. F1000Research 2021, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Clausen, P.T.L.C.; Aarestrup, F.M.; Lund, O. Rapid and Precise Alignment of Raw Reads against Redundant Databases with KMA. BMC Bioinform. 2018, 19, 307. [Google Scholar] [CrossRef] [PubMed]
- Albanese, D.; Donati, C. Strain Profiling and Epidemiology of Bacterial Species from Metagenomic Sequencing. Nat. Commun. 2017, 8, 2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-Access Bacterial Population Genomics: BIGSdb Software, the PubMLST.Org Website and Their Applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Rossi, M.; Da Silva, M.S.; Ribeiro-Gonçalves, B.F.; Silva, D.N.; Machado, M.P.; Oleastro, M.; Borges, V.; Isidro, J.; Viera, L.; Barker, D.O.R.; et al. INNUENDO Whole Genome and Core Genome MLST Schemas and Datasets for Campylobacter jejuni (1.0) [Data set]. Zenodo. 2018. Available online: https://doi.org/10.5281/zenodo.1322564 (accessed on 15 April 2022).
- Cody, A.J.; Bray, J.E.; Jolley, K.A.; McCarthy, N.D.; Maiden, M.C.J. Core Genome Multilocus Sequence Typing Scheme for Stable, Comparative Analyses of Campylobacter jejuni and C. coli Human Disease Isolates. J. Clin. Microbiol. 2017, 55, 2086–2097. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.T.; Irber, L. Sourmash: A Library for MinHash Sketching of DNA. J. Open Source Softw. 2016, 1, 27. [Google Scholar] [CrossRef] [Green Version]
- Schubert, E.; Rousseeuw, P.J. Fast and Eager k-medoids Clustering: O(k) Runtime Improvement of the pam, clara, and clarans Algorithms. Inf. Syst. 2021, 101, 101804. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.r-project.org/ (accessed on 30 December 2022).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef] [Green Version]
- Patil, I. Visualizations with Statistical Details: The “ggstatsplot” Approach. J. Open Source Softw. 2021, 6, 3167. [Google Scholar] [CrossRef]
- Pedersen, T.L. Ggraph: An Implementation of Grammar of Graphics for Graphs and Networks. 2022. Available online: https://github.com/thomasp85/ggraph (accessed on 15 April 2022).
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Ramonaitė, S.; Novoslavskij, A.; Zakarienė, G.; Aksomaitienė, J.; Malakauskas, M. High Prevalence and Genetic Diversity of Campylobacter jejuni in Wild Crows and Pigeons. Curr. Microbiol. 2015, 71, 559–565. [Google Scholar] [CrossRef]
- Lees, J.A.; Harris, S.R.; Tonkin-Hill, G.; Gladstone, R.A.; Lo, S.W.; Weiser, J.N.; Corander, J.; Bentley, S.D.; Croucher, N.J. Fast and Flexible Bacterial Genomic Epidemiology with PopPUNK. Genome Res. 2019, 29, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P.L.; et al. MetaFlye: Scalable Long-Read Metagenome Assembly Using Repeat Graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef]
- Gilpin, B.J.; Walker, T.; Paine, S.; Sherwood, J.; Mackereth, G.; Wood, T.; Hambling, T.; Hewison, C.; Brounts, A.; Wilson, M.; et al. A Large Scale Waterborne Campylobacteriosis Outbreak, Havelock North, New Zealand. J. Infect. 2020, 81, 390–395. [Google Scholar] [CrossRef]
- Izquierdo-Lara, R.W.; Elsinga, G.; Heijnen, L.; Oude Munnink, B.B.; Schapendonk, C.M.E.; Nieuwenhuijse, D.; Kon, M.; Lu, L.; Aarestrup, F.M.; Lycett, S.; et al. Monitoring SARS-CoV-2 Circulation and Diversity through Community Wastewater Sequencing, the Netherlands and Belgium. Emerg. Infect. Dis. 2021, 27, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Llarena, A.K.; Zhang, J.; Vehkala, M.; Välimäki, N.; Hakkinen, M.; Hänninen, M.L.; Roasto, M.; Mäesaar, M.; Taboada, E.; Barker, D.; et al. Monomorphic Genotypes within a Generalist Lineage of Campylobacter jejuni Show Signs of Global Dispersion. Microb. Genom. 2016, 2, e000088. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, S.K.; Cheng, L.; Méric, G.; De Haan, C.P.A.; Llarena, A.K.; Marttinen, P.; Vidal, A.; Ridley, A.; Clifton-Hadley, F.; Connor, T.R.; et al. Cryptic Ecology among Host Generalist Campylobacter jejuni in Domestic Animals. Mol. Ecol. 2014, 23, 2442–2451. [Google Scholar] [CrossRef] [Green Version]
- Mäesaar, M.; Meremäe, K.; Ivanova, M.; Roasto, M. Antimicrobial Resistance and Multilocus Sequence Types of Campylobacter jejuni Isolated from Baltic Broiler Chicken Meat and Estonian Human Patients. Poult. Sci. 2018, 97, 3645–3651. [Google Scholar] [CrossRef]
- Kivistö, R.I.; Kovanen, S.; Skarp-De Haan, A.; Schott, T.; Rahkio, M.; Rossi, M.; Hänninen, M.L. Evolutionand Comparative Genomics of Campylobacter jejuni ST-677 Clonal Complex. Genome Biol. Evol. 2014, 6, 2424–2438. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Luo, J.; Huang, J.; Wang, C.; Li, M.; Wang, B.; Wang, B.; Chang, H.; Ji, J.; Sen, K.; et al. Emergence of Genetic Diversity and Multi-Drug Resistant Campylobacter jejuni from Wild Birds in Beijing, China. Front. Microbiol. 2019, 10, 2433. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild Bird | Surface Water | |
|---|---|---|
| Number of samples analyzed | 87 | 14 |
| C. jejuni isolation positive samples | 79 | 4 |
| C. jejuni metagenomically predicted positive samples | 86 (72 *) | 13 |
| Number of isolated C. jejuni strains | 90 | 4 |
| Number of predicted strains (Europe database) | 150 | 26 |
| Number of predicted strains (WI-database) | 159 | 31 |
| Number of predicted strains (NI-database) | 143 | 32 |
| ST | #Profiles | Unique 9MLST Profiles | Profiles from Isolates | Migratory Birds/Sedentary Birds | Earliest Sampling Date | Latest Sampling Date |
|---|---|---|---|---|---|---|
| 383 | 12 | 1 | 6 | 3/9 | 2 July 2019 | 7 June 2021 |
| 45 | 9 | 4 | 6 | 1/5 | 7 May 2019 | 25 May 2021 |
| 677 | 7 | 1 | 4 | 2/5 | 7 May 2019 | 21 June 2021 |
| 11379 | 6 | 1 | 2 | 0/6 | 11 June 2019 | 7 June 2021 |
| 11383 | 5 | 1 | 1 | 0/5 | 5 May 2019 | 7 June 2021 |
| 19 | 5 | 3 | 4 | 1/3 | 18 June 2019 | 7 June 2021 |
| 448 | 5 | 3 | 4 | 0/5 | 7 May 2019 | 21 June 2021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herold, M.; Hock, L.; Penny, C.; Walczak, C.; Djabi, F.; Cauchie, H.-M.; Ragimbeau, C. Metagenomic Strain-Typing Combined with Isolate Sequencing Provides Increased Resolution of the Genetic Diversity of Campylobacter jejuni Carriage in Wild Birds. Microorganisms 2023, 11, 121. https://doi.org/10.3390/microorganisms11010121
Herold M, Hock L, Penny C, Walczak C, Djabi F, Cauchie H-M, Ragimbeau C. Metagenomic Strain-Typing Combined with Isolate Sequencing Provides Increased Resolution of the Genetic Diversity of Campylobacter jejuni Carriage in Wild Birds. Microorganisms. 2023; 11(1):121. https://doi.org/10.3390/microorganisms11010121
Chicago/Turabian StyleHerold, Malte, Louise Hock, Christian Penny, Cécile Walczak, Fatu Djabi, Henry-Michel Cauchie, and Catherine Ragimbeau. 2023. "Metagenomic Strain-Typing Combined with Isolate Sequencing Provides Increased Resolution of the Genetic Diversity of Campylobacter jejuni Carriage in Wild Birds" Microorganisms 11, no. 1: 121. https://doi.org/10.3390/microorganisms11010121