
A Comparative Analysis of the Core Proteomes within and among the Bacillus subtilis and Bacillus cereus Evolutionary Groups Reveals the Patterns of Lineage- and Species-Specific Adaptations

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analyzed Genomes
2.2. Orthology Detection and Phylogenomic Analysis
2.3. Detection of Core Proteomes
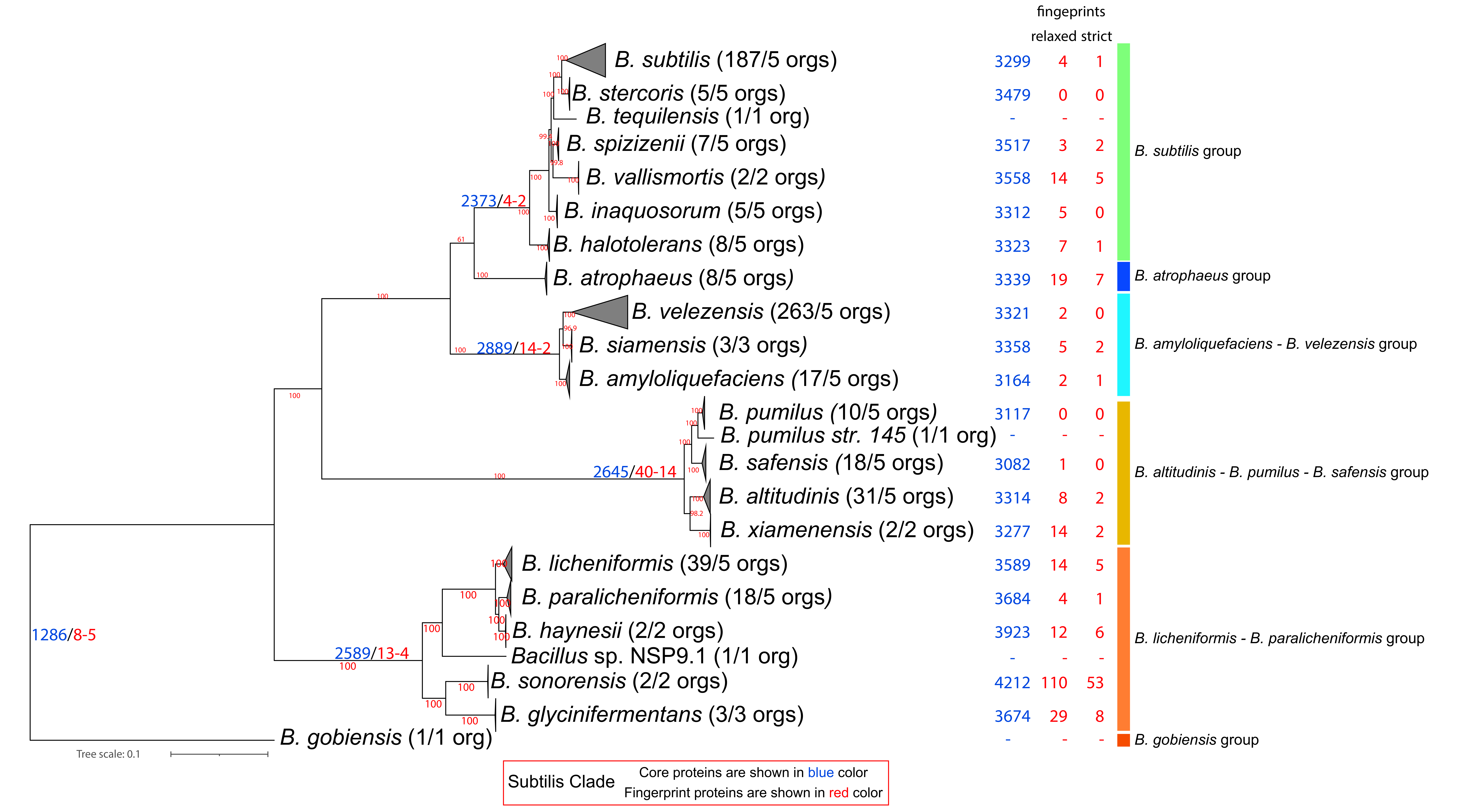
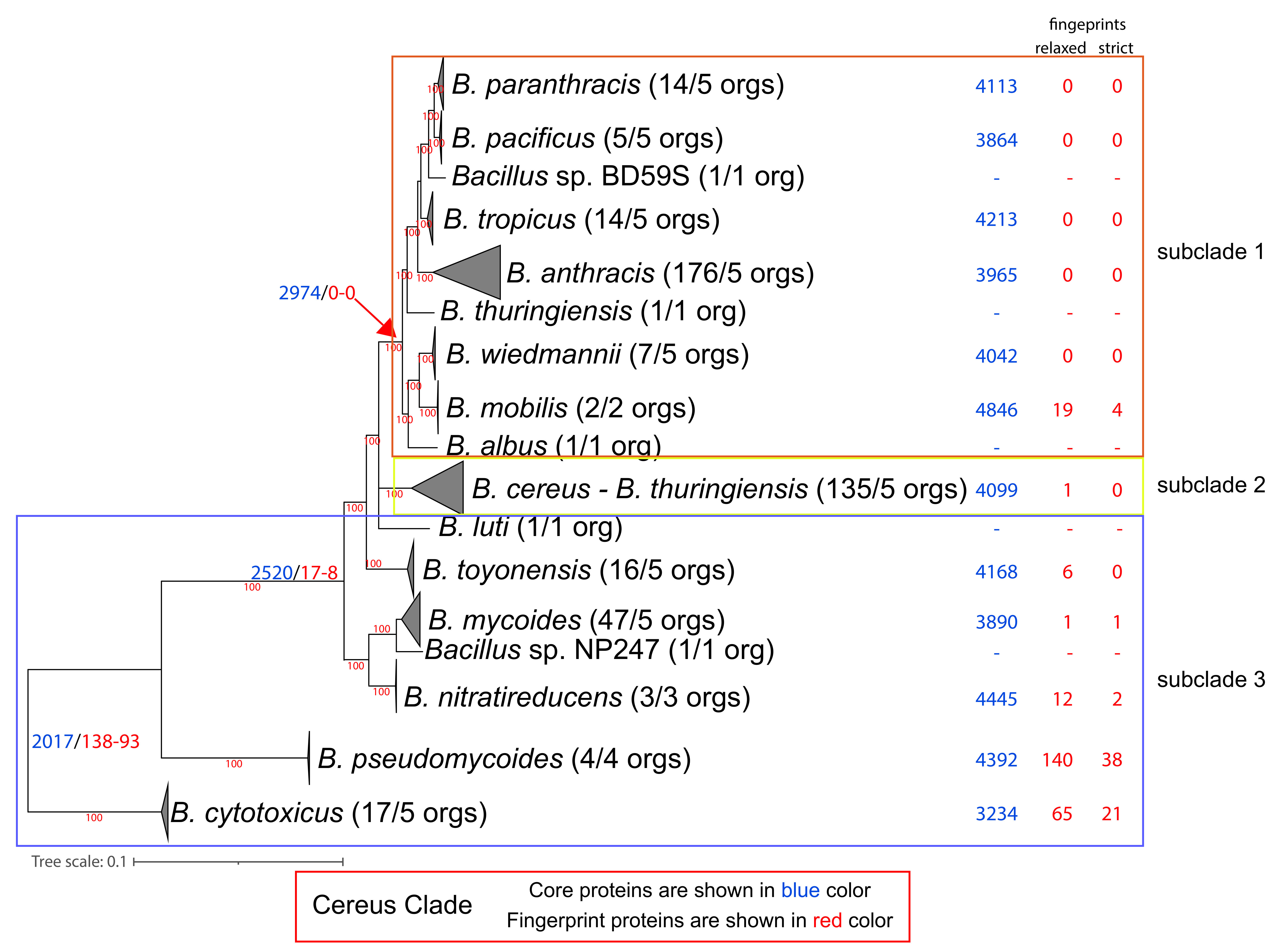
2.4. Detection of Lineage-Specific Relaxed and Strict Fingerprint Proteins
3. Results and Discussion
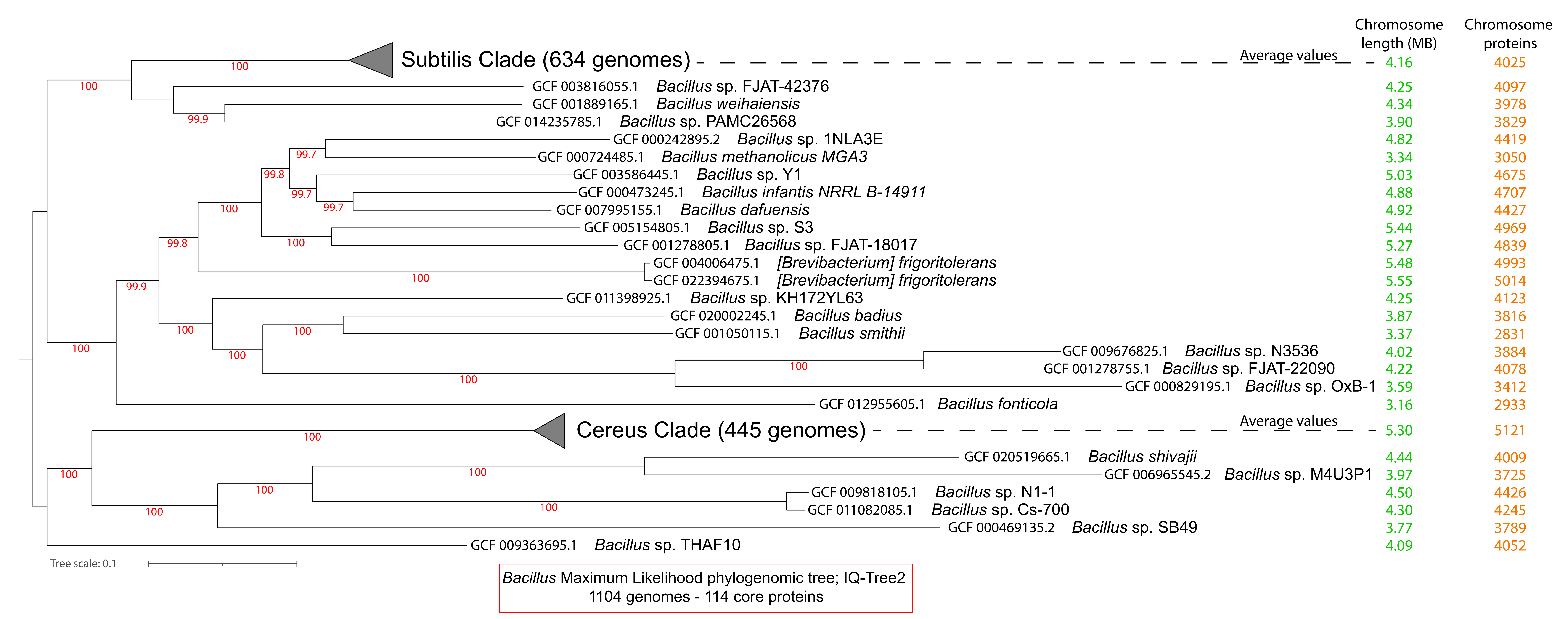
3.1. Phylogenomic Analysis of the Bacillus Genus
3.2. Phylogenomic Analysis of the Subtilis Clade
3.3. Phylogenomic Analysis of the Cereus Clade
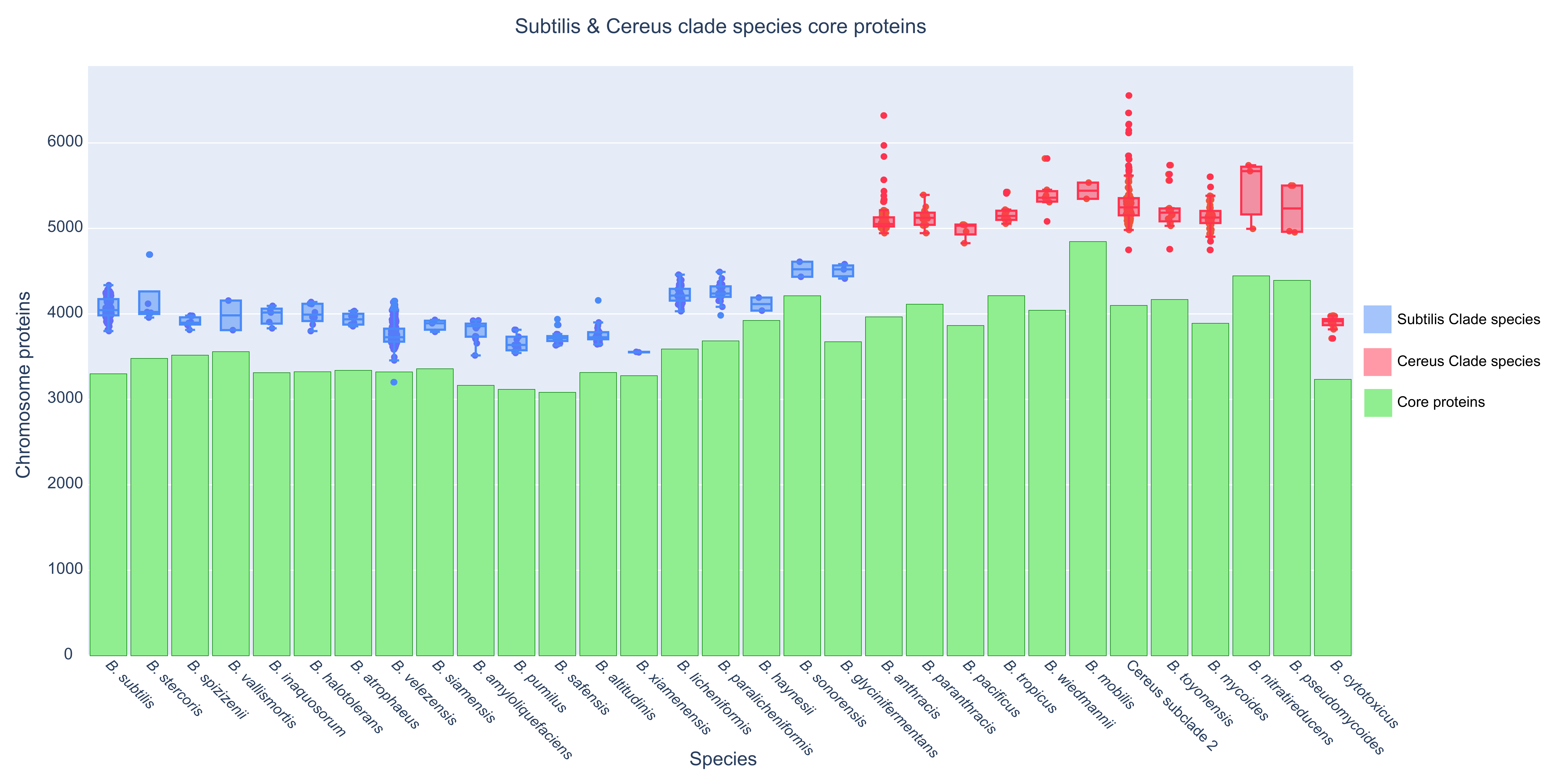
3.4. The Core Proteome and the General Genomic Characteristics of the Genus and Its Species
3.5. The Phylogenetic Distribution Profile of the Core and Accessory Protein Components of the Subtilis and Cereus Clades
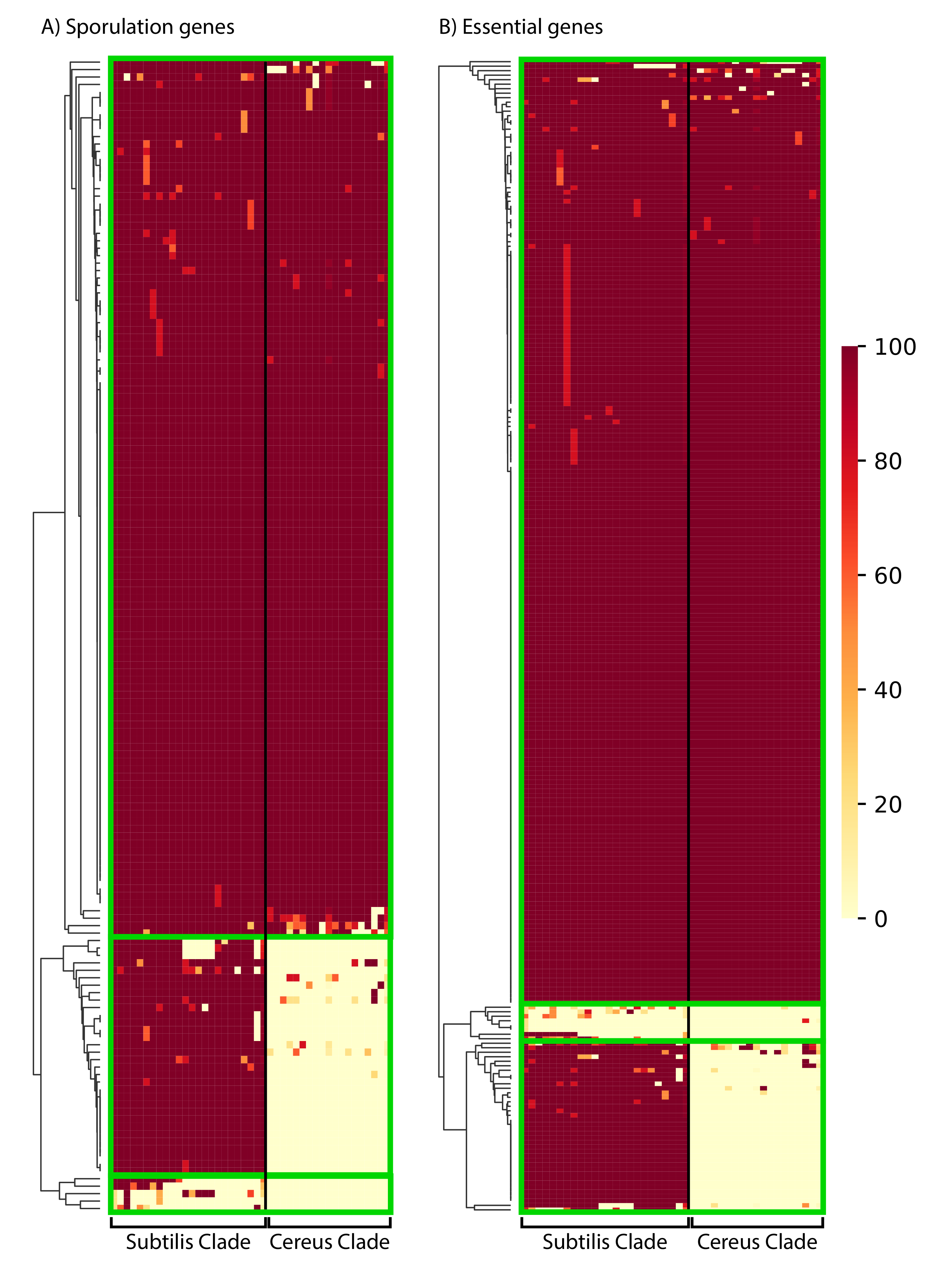
3.6. The Phylogenetic Distribution Profile of Sporulation and Essential Proteins of the Model Organism Bacillus Subtilis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fritze, D. Taxonomy of the Genus Bacillus and Related Genera: The Aerobic Endospore-Forming Bacteria. Phytopathology 2004, 94, 1245–1248. [Google Scholar] [CrossRef]
- Harwood, C.R. Introduction to the Biotechnology of Bacillus. In Bacillus; Harwood, C.R., Ed.; Springer US: Boston, MA, USA, 1989; pp. 1–4. ISBN 978-1-4899-3504-5. [Google Scholar]
- Hernández-González, I.L.; Moreno-Hagelsieb, G.; Olmedo-Álvarez, G. Environmentally-Driven Gene Content Convergence and the Bacillus Phylogeny. BMC Evol. Biol. 2018, 18, 148. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Gupta, R.S. A Phylogenomic and Comparative Genomic Framework for Resolving the Polyphyly of the Genus Bacillus: Proposal for Six New Genera of Bacillus Species, Peribacillus Gen. Nov., Cytobacillus Gen. Nov., Mesobacillus Gen. Nov., Neobacillus Gen. Nov., Metabacillus Gen. Nov. and Alkalihalobacillus Gen. Nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 406–438. [Google Scholar] [CrossRef] [PubMed]
- Ehling-Schulz, M.; Koehler, T.M.; Lereclus, D. The Bacillus cereus Group: Bacillus Species with Pathogenic Potential. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, R.; Hashem, A.; Abd Allah, E.F. Bacillus: A Biological Tool for Crop Improvement through Bio-Molecular Changes in Adverse Environments. Front. Physiol. 2017, 8, 667. [Google Scholar] [CrossRef]
- Penha, R.O.; Vandenberghe, L.P.S.; Faulds, C.; Soccol, V.T.; Soccol, C.R. Bacillus lipopeptides as Powerful Pest Control Agents for a More Sustainable and Healthy Agriculture: Recent Studies and Innovations. Planta 2020, 251, 70. [Google Scholar] [CrossRef]
- Sumi, C.D.; Yang, B.W.; Yeo, I.-C.; Hahm, Y.T. Antimicrobial Peptides of the Genus Bacillus: A New Era for Antibiotics. Can. J. Microbiol. 2015, 61, 93–103. [Google Scholar] [CrossRef]
- Mingmongkolchai, S.; Panbangred, W. Bacillus Probiotics: An Alternative to Antibiotics for Livestock Production. J. Appl. Microbiol. 2018, 124, 1334–1346. [Google Scholar] [CrossRef]
- Barbe, V.; Cruveiller, S.; Kunst, F.; Lenoble, P.; Meurice, G.; Sekowska, A.; Vallenet, D.; Wang, T.; Moszer, I.; Médigue, C.; et al. From a Consortium Sequence to a Unified Sequence: The Bacillus subtilis 168 Reference Genome a Decade Later. Microbiology 2009, 155, 1758–1775. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, L.; Li, J.; Du, G.; Chen, J. Synthetic Biology Toolbox and Chassis Development in Bacillus subtilis. Trends Biotechnol. 2019, 37, 548–562. [Google Scholar] [CrossRef]
- Parte, A.C. LPSN—List of Prokaryotic Names with Standing in Nomenclature (Bacterio.Net), 20 Years On. Int. J. Syst. Evol. Microbiol. 2018, 68, 1825–1829. [Google Scholar] [CrossRef] [PubMed]
- Logan, N.A.; Berge, O.; Bishop, A.H.; Busse, H.-J.; De Vos, P.; Fritze, D.; Heyndrickx, M.; Kämpfer, P.; Rabinovitch, L.; Salkinoja-Salonen, M.S.; et al. Proposed Minimal Standards for Describing New Taxa of Aerobic, Endospore-Forming Bacteria. Int. J. Syst. Evol. Microbiol. 2009, 59, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Côté, J.-C. Phylogenetic Relationships between Bacillus Species and Related Genera Inferred from Comparison of 3’ End 16S RDNA and 5’ End 16S-23S ITS Nucleotide Sequences. Int. J. Syst. Evol. Microbiol. 2003, 53, 695–704. [Google Scholar] [CrossRef]
- Amoutzias, G.D.; Nikolaidis, M.; Hesketh, A. The Notable Achievements and the Prospects of Bacterial Pathogen Genomics. Microorganisms 2022, 10, 1040. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hugenholtz, P.; Mavromatis, K.; Pukall, R.; Dalin, E.; Ivanova, N.N.; Kunin, V.; Goodwin, L.; Wu, M.; Tindall, B.J.; et al. A Phylogeny-Driven Genomic Encyclopaedia of Bacteria and Archaea. Nature 2009, 462, 1056–1060. [Google Scholar] [CrossRef]
- Kyrpides, N.C.; Hugenholtz, P.; Eisen, J.A.; Woyke, T.; Göker, M.; Parker, C.T.; Amann, R.; Beck, B.J.; Chain, P.S.G.; Chun, J.; et al. Genomic Encyclopedia of Bacteria and Archaea: Sequencing a Myriad of Type Strains. PLoS Biol. 2014, 12, e1001920. [Google Scholar] [CrossRef]
- Whitman, W.B.; Woyke, T.; Klenk, H.-P.; Zhou, Y.; Lilburn, T.G.; Beck, B.J.; De Vos, P.; Vandamme, P.; Eisen, J.A.; Garrity, G.; et al. Genomic Encyclopedia of Bacterial and Archaeal Type Strains, Phase III: The Genomes of Soil and Plant-Associated and Newly Described Type Strains. Stand. Genom. Sci. 2015, 10, 26. [Google Scholar] [CrossRef]
- Kunin, V.; Goldovsky, L.; Darzentas, N.; Ouzounis, C.A. The Net of Life: Reconstructing the Microbial Phylogenetic Network. Genome Res. 2005, 15, 954–959. [Google Scholar] [CrossRef]
- Kunin, V.; Ouzounis, C.A. The Balance of Driving Forces during Genome Evolution in Prokaryotes. Genome Res. 2003, 13, 1589–1594. [Google Scholar] [CrossRef]
- Gogarten, J.P.; Townsend, J.P. Horizontal Gene Transfer, Genome Innovation and Evolution. Nat. Rev. Microbiol. 2005, 3, 679–687. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome Analysis of Multiple Pathogenic Isolates of Streptococcus Agalactiae: Implications for the Microbial “Pan-Genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten Years of Pan-Genome Analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- Jun, S.-R.; Wassenaar, T.M.; Nookaew, I.; Hauser, L.; Wanchai, V.; Land, M.; Timm, C.M.; Lu, T.-Y.S.; Schadt, C.W.; Doktycz, M.J.; et al. Diversity of Pseudomonas Genomes, Including Populus-Associated Isolates, as Revealed by Comparative Genome Analysis. Appl. Environ. Microbiol. 2016, 82, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic Insights That Advance the Species Definition for Prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the Genomic Gold Standard for the Prokaryotic Species Definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Zwick, M.E.; Joseph, S.J.; Didelot, X.; Chen, P.E.; Bishop-Lilly, K.A.; Stewart, A.C.; Willner, K.; Nolan, N.; Lentz, S.; Thomason, M.K.; et al. Genomic Characterization of the Bacillus cereus Sensu Lato Species: Backdrop to the Evolution of Bacillus anthracis. Genome Res. 2012, 22, 1512–1524. [Google Scholar] [CrossRef]
- Bazinet, A.L. Pan-Genome and Phylogeny of Bacillus Cereus Sensu Lato. BMC Evol. Biol. 2017, 17, 176. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, J.; Lai, Q.; Zeng, R.; Ye, D.; Xu, J.; Shao, Z. Proposal of Nine Novel Species of the Bacillus Cereus Group. Int. J. Syst. Evol. Microbiol. 2017, 67, 2499–2508. [Google Scholar] [CrossRef]
- Chang, T.; Rosch, J.W.; Gu, Z.; Hakim, H.; Hewitt, C.; Gaur, A.; Wu, G.; Hayden, R.T. Whole-Genome Characterization of Bacillus cereus Associated with Specific Disease Manifestations. Infect. Immun. 2018, 86, e00574-17. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Blom, J.; Klenk, H.-P.; Borriss, R. Bacillus amyloliquefaciens, Bacillus velezensis, and Bacillus siamensis Form an “Operational Group B. Amyloliquefaciens” within the B. Subtilis Species Complex. Front. Microbiol. 2017, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Tirumalai, M.R.; Stepanov, V.G.; Wünsche, A.; Montazari, S.; Gonzalez, R.O.; Venkateswaran, K.; Fox, G.E. Bacillus safensis FO-36b and Bacillus pumilus SAFR-032: A Whole Genome Comparison of Two Spacecraft Assembly Facility Isolates. BMC Microbiol. 2018, 18, 57. [Google Scholar] [CrossRef]
- Du, Y.; Ma, J.; Yin, Z.; Liu, K.; Yao, G.; Xu, W.; Fan, L.; Du, B.; Ding, Y.; Wang, C. Comparative Genomic Analysis of Bacillus paralicheniformis MDJK30 with Its Closely Related Species Reveals an Evolutionary Relationship between B. paralicheniformis and B. licheniformis. BMC Genom. 2019, 20, 283. [Google Scholar] [CrossRef]
- Wu, H.; Wang, D.; Gao, F. Toward a High-Quality Pan-Genome Landscape of Bacillus subtilis by Removal of Confounding Strains. Brief. Bioinform. 2021, 22, 1951–1971. [Google Scholar] [CrossRef] [PubMed]
- Dunlap, C.A.; Bowman, M.J.; Zeigler, D.R. Promotion of Bacillus Subtilis Subsp. Inaquosorum, Bacillus subtilis Subsp. spizizenii and Bacillus subtilis Subsp. stercoris to Species Status. Antonie Van Leeuwenhoek 2020, 113, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.J.A.; Tasara, T.; Klumpp, J.; Stephan, R.; Ehling-Schulz, M.; Johler, S. Whole-Genome-Based Phylogeny of Bacillus cytotoxicus Reveals Different Clades within the Species and Provides Clues on Ecology and Evolution. Sci. Rep. 2019, 9, 1984. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Patel, S.; Saini, N.; Chen, S. Robust Demarcation of 17 Distinct Bacillus Species Clades, Proposed as Novel Bacillaceae Genera, by Phylogenomics and Comparative Genomic Analyses: Description of Robertmurraya kyonggiensis sp. Nov. and Proposal for an Emended Genus Bacillus Limiting It Only to the Members of the Subtilis and Cereus Clades of Species. Int. J. Syst. Evol. Microbiol. 2020, 70, 5753–5798. [Google Scholar] [CrossRef]
- Bhandari, V.; Ahmod, N.Z.; Shah, H.N.; Gupta, R.S. Molecular Signatures for Bacillus Species: Demarcation of the Bacillus subtilis and Bacillus cereus Clades in Molecular Terms and Proposal to Limit the Placement of New Species into the Genus Bacillus. Int. J. Syst. Evol. Microbiol. 2013, 63, 2712–2726. [Google Scholar] [CrossRef]
- Parker, C.T.; Tindall, B.J.; Garrity, G. International Code of Nomenclature of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2019, 69, S1–S111. [Google Scholar] [CrossRef]
- Nikolaidis, M.; Mossialos, D.; Oliver, S.G.; Amoutzias, G.D. Comparative Analysis of the Core Proteomes among the Pseudomonas Major Evolutionary Groups Reveals Species-Specific Adaptations for Pseudomonas aeruginosa and Pseudomonas chlororaphis. Diversity 2020, 12, 289. [Google Scholar] [CrossRef]
- Bentley, S.D.; Vernikos, G.S.; Snyder, L.A.S.; Churcher, C.; Arrowsmith, C.; Chillingworth, T.; Cronin, A.; Davis, P.H.; Holroyd, N.E.; Jagels, K.; et al. Meningococcal Genetic Variation Mechanisms Viewed through Comparative Analysis of Serogroup C Strain FAM18. PLoS Genet. 2007, 3, e23. [Google Scholar] [CrossRef] [Green Version]
- Hiller, N.L.; Janto, B.; Hogg, J.S.; Boissy, R.; Yu, S.; Powell, E.; Keefe, R.; Ehrlich, N.E.; Shen, K.; Hayes, J.; et al. Comparative Genomic Analyses of Seventeen Streptococcus pneumoniae Strains: Insights into the Pneumococcal Supragenome. J. Bacteriol. 2007, 189, 8186–8195. [Google Scholar] [CrossRef] [PubMed]
- Méric, G.; Yahara, K.; Mageiros, L.; Pascoe, B.; Maiden, M.C.J.; Jolley, K.A.; Sheppard, S.K. A Reference Pan-Genome Approach to Comparative Bacterial Genomics: Identification of Novel Epidemiological Markers in Pathogenic Campylobacter. PLoS ONE 2014, 9, e92798. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Rosovitz, M.J.; Myers, G.S.A.; Mongodin, E.F.; Fricke, W.F.; Gajer, P.; Crabtree, J.; Sebaihia, M.; Thomson, N.R.; Chaudhuri, R.; et al. The Pangenome Structure of Escherichia coli: Comparative Genomic Analysis of E. coli Commensal and Pathogenic Isolates. J. Bacteriol. 2008, 190, 6881–6893. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Chevenet, F.; Brun, C.; Bañuls, A.-L.; Jacq, B.; Christen, R. TreeDyn: Towards Dynamic Graphics and Annotations for Analyses of Trees. BMC Bioinform. 2006, 7, 439. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v4: Recent Updates and New Developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and Taxonomy in Diagnostics for Food Security: Soft-Rotting Enterobacterial Plant Pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An Automatic Genome Annotation and Pathway Reconstruction Server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded Microbial Genome Coverage and Improved Protein Family Annotation in the COG Database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef]
- Federhen, S. Type Material in the NCBI Taxonomy Database. Nucleic Acids Res. 2015, 43, D1086–D1098. [Google Scholar] [CrossRef]
- Anagnostopoulos, C.; Spizizen, J. Requirements for transformation in Bacillus subtilis. J. Bacteriol. 1961, 81, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Kunst, F.; Ogasawara, N.; Moszer, I.; Albertini, A.M.; Alloni, G.; Azevedo, V.; Bertero, M.G.; Bessières, P.; Bolotin, A.; Borchert, S.; et al. The Complete Genome Sequence of the Gram-Positive Bacterium Bacillus subtilis. Nature 1997, 390, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Borriss, R.; Danchin, A.; Harwood, C.R.; Médigue, C.; Rocha, E.P.C.; Sekowska, A.; Vallenet, D. Bacillus subtilis, the Model Gram-Positive Bacterium: 20 Years of Annotation Refinement. Microb. Biotechnol. 2018, 11, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Rooney, A.P.; Price, N.P.J.; Ehrhardt, C.; Swezey, J.L.; Bannan, J.D. Phylogeny and Molecular Taxonomy of the Bacillus subtilis Species Complex and Description of Bacillus subtilis Subsp. inaquosorum Subsp. Nov. Int. J. Syst. Evol. Microbiol. 2009, 59, 2429–2436. [Google Scholar] [CrossRef]
- Guinebretière, M.-H.; Auger, S.; Galleron, N.; Contzen, M.; De Sarrau, B.; De Buyser, M.-L.; Lamberet, G.; Fagerlund, A.; Granum, P.E.; Lereclus, D.; et al. Bacillus cytotoxicus Sp. Nov. Is a Novel Thermotolerant Species of the Bacillus cereus Group Occasionally Associated with Food Poisoning. Int. J. Syst. Evol. Microbiol. 2013, 63, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lai, Q.; Göker, M.; Meier-Kolthoff, J.P.; Wang, M.; Sun, Y.; Wang, L.; Shao, Z. Genomic Insights into the Taxonomic Status of the Bacillus cereus Group. Sci. Rep. 2015, 5, 14082. [Google Scholar] [CrossRef] [Green Version]
- Baek, I.; Lee, K.; Goodfellow, M.; Chun, J. Comparative Genomic and Phylogenomic Analyses Clarify Relationships within and Between Bacillus cereus and Bacillus thuringiensis: Proposal for the Recognition of Two Bacillus thuringiensis Genomovars. Front. Microbiol. 2019, 10, 1978. [Google Scholar] [CrossRef]
- Sorokin, A.; Candelon, B.; Guilloux, K.; Galleron, N.; Wackerow-Kouzova, N.; Ehrlich, S.D.; Bourguet, D.; Sanchis, V. Multiple-Locus Sequence Typing Analysis of Bacillus cereus and Bacillus thuringiensis Reveals Separate Clustering and a Distinct Population Structure of Psychrotrophic Strains. Appl. Environ. Microbiol. 2006, 72, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Barker, M.; Falush, D.; Priest, F.G. Evolution of Pathogenicity in the Bacillus cereus Group. Syst. Appl. Microbiol. 2009, 32, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Okinaka, R.T.; Keim, P. The Phylogeny of Bacillus cereus sensu lato. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, R. Some aspects of evolution. Science 1933, 78, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Kolstø, A.-B.; Tourasse, N.J.; Økstad, O.A. What Sets Bacillus anthracis Apart from Other Bacillus Species? Annu. Rev. Microbiol. 2009, 63, 451–476. [Google Scholar] [CrossRef]
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax Pathogenesis. Annu. Rev. Microbiol. 2015, 69, 185–208. [Google Scholar] [CrossRef]
- Helgason, E.; Okstad, O.A.; Caugant, D.A.; Johansen, H.A.; Fouet, A.; Mock, M.; Hegna, I.; Kolstø, A.B. Bacillus Anthracis, Bacillus Cereus, and Bacillus Thuringiensis--One Species on the Basis of Genetic Evidence. Appl. Environ. Microbiol. 2000, 66, 2627–2630. [Google Scholar] [CrossRef]
- Rasko, D.A.; Altherr, M.R.; Han, C.S.; Ravel, J. Genomics of the Bacillus cereus Group of Organisms. FEMS Microbiol. Rev. 2005, 29, 303–329. [Google Scholar] [CrossRef]
- Patiño-Navarrete, R.; Sanchis, V. Evolutionary Processes and Environmental Factors Underlying the Genetic Diversity and Lifestyles of Bacillus cereus Group Bacteria. Res. Microbiol. 2017, 168, 309–318. [Google Scholar] [CrossRef]
- Rasigade, J.-P.; Hollandt, F.; Wirth, T. Genes under Positive Selection in the Core Genome of Pathogenic Bacillus cereus Group Members. Infect. Genet. Evol. 2018, 65, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, N.; Sorokin, A.; Anderson, I.; Galleron, N.; Candelon, B.; Kapatral, V.; Bhattacharyya, A.; Reznik, G.; Mikhailova, N.; Lapidus, A.; et al. Genome Sequence of Bacillus cereus and Comparative Analysis with Bacillus anthracis. Nature 2003, 423, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Read, T.D.; Peterson, S.N.; Tourasse, N.; Baillie, L.W.; Paulsen, I.T.; Nelson, K.E.; Tettelin, H.; Fouts, D.E.; Eisen, J.A.; Gill, S.R.; et al. The Genome Sequence of Bacillus anthracis Ames and Comparison to Closely Related Bacteria. Nature 2003, 423, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Margulis, L.; Jorgensen, J.Z.; Dolan, S.; Kolchinsky, R.; Rainey, F.A.; Lo, S.C. The Arthromitus Stage of Bacillus cereus: Intestinal Symbionts of Animals. Proc. Natl. Acad. Sci. USA 1998, 95, 1236–1241. [Google Scholar] [CrossRef]
- Wood, V.; Lock, A.; Harris, M.A.; Rutherford, K.; Bähler, J.; Oliver, S.G. Hidden in Plain Sight: What Remains to Be Discovered in the Eukaryotic Proteome? Open Biol. 2019, 9, 180241. [Google Scholar] [CrossRef]
- Kim, Y.; Koh, I.; Young Lim, M.; Chung, W.-H.; Rho, M. Pan-Genome Analysis of Bacillus for Microbiome Profiling. Sci. Rep. 2017, 7, 10984. [Google Scholar] [CrossRef]
- Link, L.; Sawyer, J.; Venkateswaran, K.; Nicholson, W. Extreme Spore UV Resistance of Bacillus pumilus Isolates Obtained from an Ultraclean Spacecraft Assembly Facility. Microb. Ecol. 2004, 47, 159–163. [Google Scholar] [CrossRef]
- Satomi, M.; La Duc, M.T.; Venkateswaran, K. Bacillus safensis Sp. Nov., Isolated from Spacecraft and Assembly-Facility Surfaces. Int. J. Syst. Evol. Microbiol. 2006, 56, 1735–1740. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Mekhedov, S.L.; Puigbo, P.; Smirnov, S.; Wolf, Y.I.; Rigden, D.J. Genomic Determinants of Sporulation in Bacilli and Clostridia: Towards the Minimal Set of Sporulation-Specific Genes. Environ. Microbiol. 2012, 14, 2870–2890. [Google Scholar] [CrossRef]
- Eijlander, R.T.; de Jong, A.; Krawczyk, A.O.; Holsappel, S.; Kuipers, O.P. SporeWeb: An Interactive Journey through the Complete Sporulation Cycle of Bacillus subtilis. Nucleic Acids Res. 2014, 42, D685–D691. [Google Scholar] [CrossRef] [Green Version]
- Michna, R.H.; Zhu, B.; Mäder, U.; Stülke, J. SubtiWiki 2.0—An Integrated Database for the Model Organism Bacillus subtilis. Nucleic Acids Res. 2016, 44, D654–D662. [Google Scholar] [CrossRef] [PubMed]
- Sella, S.R.B.R.; Vandenberghe, L.P.S.; Soccol, C.R. Life Cycle and Spore Resistance of Spore-Forming Bacillus atrophaeus. Microbiol. Res. 2014, 169, 931–939. [Google Scholar] [CrossRef] [PubMed]
- McKenney, P.T.; Driks, A.; Eichenberger, P. The Bacillus subtilis Endospore: Assembly and Functions of the Multilayered Coat. Nat. Rev. Microbiol. 2013, 11, 33–44. [Google Scholar] [CrossRef]
- Higgins, D.; Dworkin, J. Recent Progress in Bacillus subtilis Sporulation. FEMS Microbiol. Rev. 2012, 36, 131–148. [Google Scholar] [CrossRef]
- Zhu, B.; Stülke, J. SubtiWiki in 2018: From Genes and Proteins to Functional Network Annotation of the Model Organism Bacillus subtilis. Nucleic Acids Res. 2018, 46, D743–D748. [Google Scholar] [CrossRef]
- Meeske, A.J.; Rodrigues, C.D.A.; Brady, J.; Lim, H.C.; Bernhardt, T.G.; Rudner, D.Z. High-Throughput Genetic Screens Identify a Large and Diverse Collection of New Sporulation Genes in Bacillus subtilis. PLoS Biol. 2016, 14, e1002341. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxonomy | Num. Genomes | Avg. Chrom. Length (MB) | Avg. Num of Chrom. Proteins | Core Proteins Normalized | Avg. Num of Accessory Proteins | Relaxed Fingerprints | Strict Fingerprints | Fingerprints (Relaxed/Strict) with Function Unknown |
|---|---|---|---|---|---|---|---|---|
| Subtilis Clade | 630 | 4.06 | 3914 | 1286 | 2628 | 8 | 5 | 6/5 |
| B. subtilis group | 214 | 4.14 | 4061 | 2373 | 1688 | 4 | 2 | 4/2 |
| B. subtilis species | 187 | 4.14 | 4070 | 3299 | 771 | 4 | 1 | 4/1 |
| B. stercoris species | 5 | 4.29 | 4161 | 3479 | 682 | 0 | 0 | 0/0 |
| B. tequilensis species * | 1 | 4.01 | 3943 | - | - | - | - | - |
| B. spizizenii species | 7 | 4.08 | 3905 | 3517 | 388 | 3 | 2 | 3/2 |
| B. vallismortis species * | 2 | 4.12 | 3983 | 3558 | 425 | 14 | 5 | 14/5 |
| B. inaquosorum species | 5 | 4.24 | 3979 | 3312 | 667 | 5 | 0 | 5/0 |
| B. halotolerans species | 8 | 4.13 | 3999 | 3323 | 676 | 7 | 1 | 6/1 |
| B. atrophaeus group | 8 | 4.19 | 3939 | 3339 | 600 | 19 | 7 | 17/6 |
| B. amyloliquefaciens—B. velezensis group | 283 | 4.00 | 3768 | 2889 | 879 | 14 | 2 | 12/2 |
| B. amyloliquefaciens species | 17 | 3.93 | 3810 | 3164 | 646 | 2 | 1 | 2/1 |
| B. velezensis species | 263 | 4.01 | 3764 | 3321 | 443 | 2 | 0 | 2/0 |
| B. siamensis species | 3 | 4.12 | 3870 | 3358 | 512 | 5 | 2 | 3/1 |
| B. pumilus-B. safensis-B. altitudinis group | 61 | 3.77 | 3723 | 2645 | 1078 | 40 | 14 | 28/14 |
| B. pumilus species | 10 | 3.73 | 3661 | 3117 | 544 | 0 | 0 | 0/0 |
| B. altitudinis species | 31 | 3.77 | 3753 | 3314 | 439 | 8 | 2 | 8/2 |
| B. safensis species | 18 | 3.78 | 3725 | 3082 | 643 | 1 | 0 | 1/0 |
| B. xiamenensis species * | 2 | 3.63 | 3553 | 3277 | 276 | 14 | 2 | 13/2 |
| B. pumilus str. 145 * | 1 | 3.94 | 3898 | - | - | - | - | - |
| B. licheniformis—paralicheniformis group | 64 | 4.36 | 4252 | 2589 | 1663 | 13 | 4 | 7/4 |
| B. licheniformis species | 39 | 4.30 | 4228 | 3589 | 639 | 14 | 5 | 12/5 |
| B. paralicheniformis species | 18 | 4.42 | 4248 | 3684 | 564 | 4 | 1 | 4/1 |
| B. haynesii species * | 2 | 4.28 | 4115 | 3923 | 192 | 12 | 6 | 12/6 |
| Bacillus sp. NSP9.1 * | 1 | 4.54 | 4496 | - | - | - | - | - |
| B. sonorensis species * | 2 | 4.72 | 4522 | 4212 | 310 | 110 | 53 | 93/52 |
| B. glycinifermentans species | 3 | 4.67 | 4505 | 3674 | 831 | 29 | 8 | 27/8 |
| B. gobiensis group | 1 | 4.60 | 4455 | - | - | - | - | - |
| Cereus Clade | 305 | 5.24 | 5062 | 2017 | 3045 | 138 | 93 | 102/86 |
| Cereus subclade 1 | 218 | 5.28 | 5115 | 2974 | 2141 | 0 | 0 | 0/0 |
| B. paranthracis species | 14 | 5.28 | 5134 | 4113 | 1021 | 0 | 0 | 0/0 |
| B. pacificus species | 5 | 5.13 | 4984 | 3864 | 1120 | 0 | 0 | 0/0 |
| Bacillus sp. BD59S * | 1 | 5.28 | 5168 | - | - | - | - | - |
| B. tropicus species | 14 | 5.34 | 5177 | 4213 | 964 | 0 | 0 | 0/0 |
| B. anthracis species | 176 | 5.27 | 5097 | 3965 | 1132 | 0 | 0 | 0/0 |
| B. anthracis clonal clade | 111 | 5.23 | 5035 | 4881 | 154 | 45 | 15 | 37/15 |
| B. thuringiensis * | 1 | 5.33 | 5198 | - | - | - | - | - |
| B. wiedmannii species | 7 | 5.55 | 5391 | 4042 | 1349 | 0 | 0 | 0/0 |
| B. mobilis species * | 2 | 5.56 | 5441 | 4846 | 595 | 19 | 4 | 17/4 |
| B. albus species * | 1 | 5.30 | 5101 | - | - | - | - | - |
| Cereus subclade 2 | 135 | 5.53 | 5305 | 4099 | 1206 | 1 | 0 | 1/0 |
| Cereus subclades 1 & 2 | 218 | 5.28 | 5115 | 2881 | 2234 | 0 | 0 | 0/0 |
| Cereus subclade 3 | - | - | - | - | - | - | - | - |
| B. luti species * | 1 | 5.20 | 4992 | - | - | - | - | - |
| B. toyonensis species | 16 | 5.40 | 5217 | 4168 | 1049 | 6 | 0 | 4/0 |
| B. mycoides species | 47 | 5.32 | 5144 | 3890 | 1254 | 1 | 1 | 1/1 |
| Bacillus sp. NP247 * | 1 | 5.28 | 5107 | - | - | - | - | - |
| B. nitratireducens species | 3 | 5.57 | 5468 | 4445 | 1023 | 12 | 2 | 10/2 |
| B. pseudomycoides species | 4 | 5.49 | 5231 | 4392 | 839 | 140 | 38 | 101/37 |
| B. cytotoxicus species | 17 | 4.19 | 3897 | 3234 | 663 | 65 | 21 | 51/21 |
| Cereus subclade1&2-mycoides CA | 284 | 5.30 | 5129 | 2520 | 2609 | 17 | 8 | 13/8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolaidis, M.; Hesketh, A.; Mossialos, D.; Iliopoulos, I.; Oliver, S.G.; Amoutzias, G.D. A Comparative Analysis of the Core Proteomes within and among the Bacillus subtilis and Bacillus cereus Evolutionary Groups Reveals the Patterns of Lineage- and Species-Specific Adaptations. Microorganisms 2022, 10, 1720. https://doi.org/10.3390/microorganisms10091720
Nikolaidis M, Hesketh A, Mossialos D, Iliopoulos I, Oliver SG, Amoutzias GD. A Comparative Analysis of the Core Proteomes within and among the Bacillus subtilis and Bacillus cereus Evolutionary Groups Reveals the Patterns of Lineage- and Species-Specific Adaptations. Microorganisms. 2022; 10(9):1720. https://doi.org/10.3390/microorganisms10091720
Chicago/Turabian StyleNikolaidis, Marios, Andrew Hesketh, Dimitris Mossialos, Ioannis Iliopoulos, Stephen G. Oliver, and Grigorios D. Amoutzias. 2022. "A Comparative Analysis of the Core Proteomes within and among the Bacillus subtilis and Bacillus cereus Evolutionary Groups Reveals the Patterns of Lineage- and Species-Specific Adaptations" Microorganisms 10, no. 9: 1720. https://doi.org/10.3390/microorganisms10091720