
Characterization of Cronobacter sakazakii Strains Originating from Plant-Origin Foods Using Comparative Genomic Analyses and Zebrafish Infectivity Studies

 , , , add
Show full author list
, , , add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. DNA Extraction from Plant-Origin Isolates
2.3. Whole Genome Sequencing, Assemblies, Annotation, and Multi-Locus Sequence Typing
2.4. Comparative Genome Analyses: Phylogeny, Blast Analyses of Malonate/Xylose Utilization Operons
2.5. Analyses of Antimicrobial Resistance Genes, Prophage Sequence Profiles, and Clusters of Orthologous Groups
2.6. Microarray Design, Hybridization, and Analysis
2.7. Zebrafish Embryo Infection
3. Results and Discussion
3.1. Identities and Genome Properties of C. sakazakii Strains Originating from Plant-Origin Food
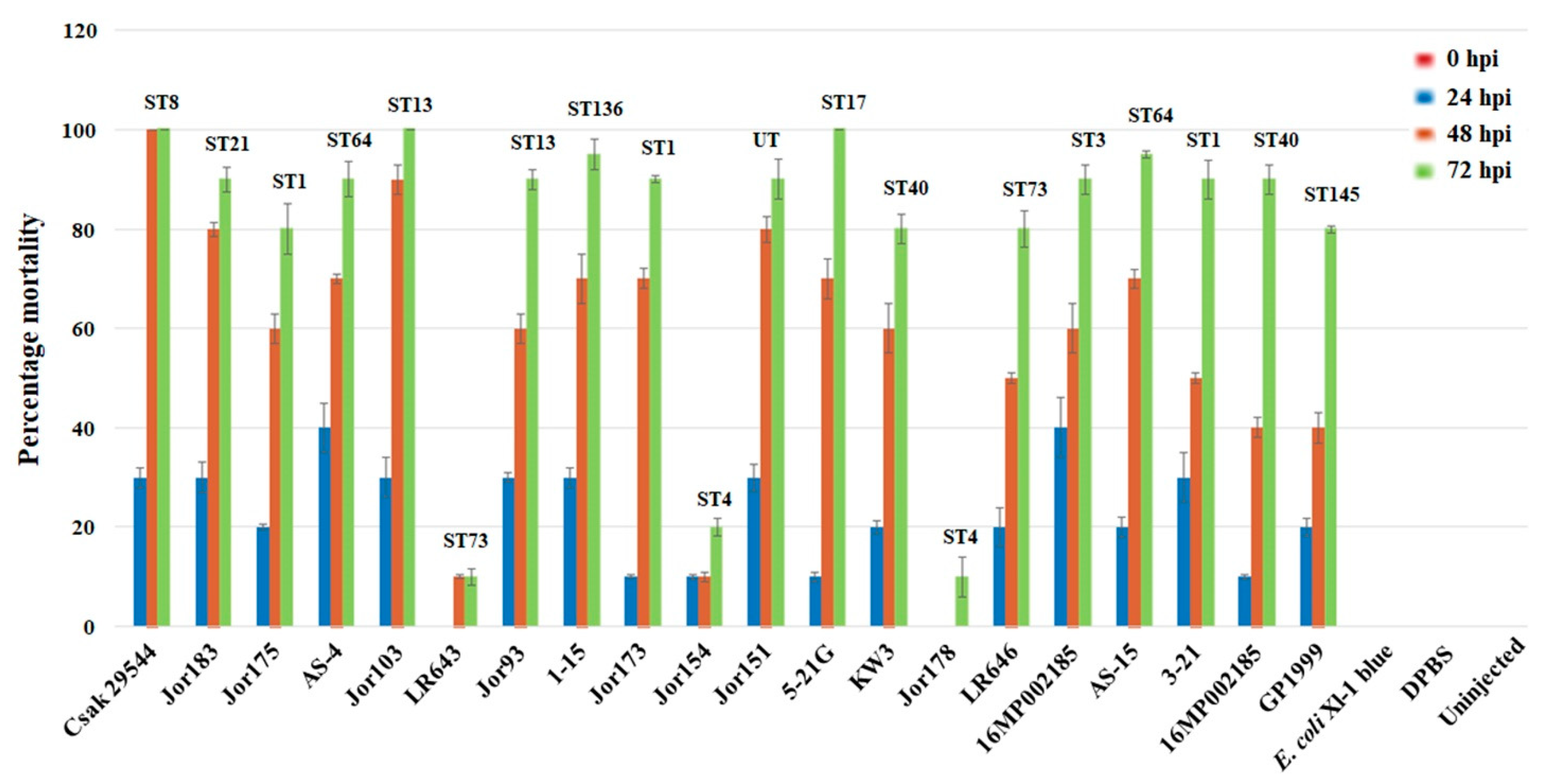
3.2. Zebrafish Embryo Infectivity Studies
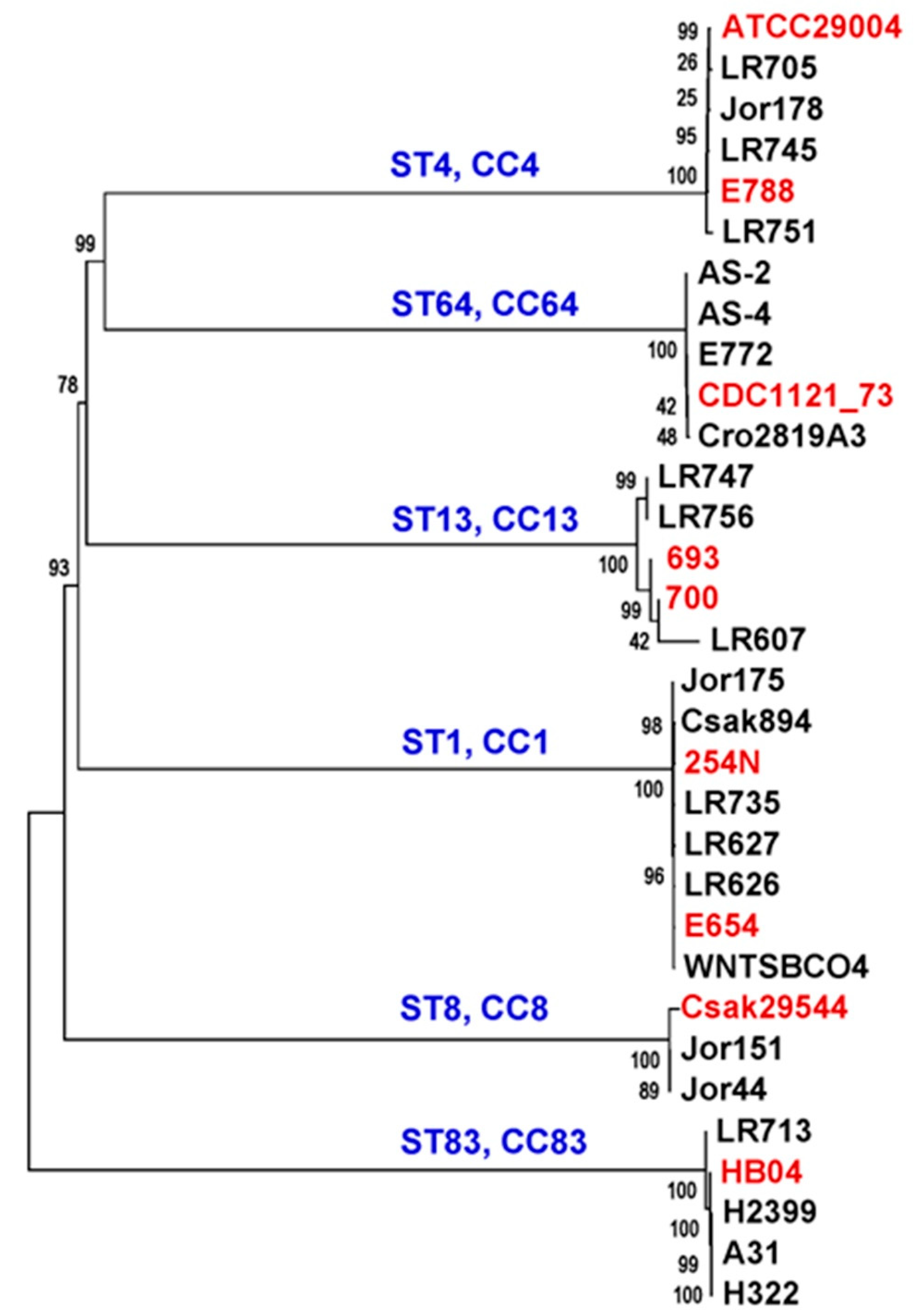
3.3. Phylogenetic Characterization of Cronobacter Strains from Plant-Origin Foods Using a Genus-Specific Core-Gene MLST Schema
3.4. Identification of Antimicrobial Resistance Genes, Prophage Signature Sequences, and Clusters of Orthologous Groups in Plant-Origin Cronobacter Strains
3.5. Prevalence and Distribution of Plasmids and Respective Virulence Factor Genes among C. sakazakii Strains Derived from Plant-Origin Foods
3.6. Prevalence and Distribution of the Xylose Utilization Operon in Cronobacter Strains Isolated from Plant-Origin Foods
3.7. DNA Tiling Microarray Analysis
3.8. Important Lessons from This Surveillance Study for Food Safety
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Iversen, C.; Mullane, N.; McCardell, B.; Tall, B.D.; Lehner, A.; Fanning, S.; Stephan, R.; Joosten, H. Cronobacter gen. nov., a new genus to accommodate the biogroups of Enterobacter sakazakii, and proposal of Cronobacter sakazakii gen. nov., comb. nov., Cronobacter malonaticus sp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov., Cronobacter genomospecies 1 and of three subspecies, Cronobacter dublinensis subsp. dublinensis subsp. nov., Cronobacter dublinensis subsp. lausannensis subsp. nov. and Cronobacter dublinensis subsp. lactaridi subsp. nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.; Cetinkaya, E.; Drahovska, H.; Levican, A.; Figueras, M.J.; Forsythe, S.J. Cronobacter condimenti sp. nov., isolated from spiced meat and Cronobacter universalis sp. nov., a species designation for Cronobacter sp. genomospecies 1, recovered from a leg infection, water and food ingredients. Int. J. Syst. Evol. Microbiol. 2012, 62, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Stephan, R.; Grim, C.J.; Gopinath, G.R.; Mammel, M.K.; Sathyamoorthy, V.; Trach, L.H.; Chase, H.R.; Fanning, S.; Tall, B.D. Re-examination of the taxonomic status of Enterobacter helveticus sp. nov., Enterobacter pulveris sp. nov., and Enterobacter turicensis sp. nov. as members of Cronobacter: Proposal of two new genera Siccibacter gen. nov. and Franconibacter gen. nov. and descriptions of Siccibacter turicensis sp. nov., Franconibacter helveticus sp. nov., and Franconibacter pulveris sp. nov. Int. J. Syst. Evol. Microbiol. 2014, 64, 3402–3410. [Google Scholar] [PubMed] [Green Version]
- Iversen, C.; Forsythe, S. Risk profile of Enterobacter sakazakii, an emerging pathogen associated with infant milk formula. Trends Food Sci. Technol. 2003, 14, 443–454. [Google Scholar] [CrossRef]
- Tall, B.D.; Chen, Y.; Yan, Q.Q.; Gopinath, G.R.; Grim, C.J.; Jarvis, K.G.; Fanning, S.; Lampel, K.A. Cronobacter: An emergent pathogen a using meningitis to neonates through their feeds. Sci. Prog. 2014, 97, 154–172. [Google Scholar] [CrossRef]
- Hunter, C.J.; Petrosyan, M.; Ford, H.R.; Prasadarao, N.V. Enterobacter sakazakii: An emerging pathogen in infants and neonates. Surg. Infect. 2008, 9, 533–539. [Google Scholar] [CrossRef]
- Holý, O.; Petrželová, J.; Hanulík, V.; Chromá, M.; Matoušková, I.; Forsythe, S.J. Epidemiology of Cronobacter spp. isolates from patients admitted to the Olomouc University Hospital (Czech Republic). Epidemiol. Mikrobiol. Imunol. 2014, 63, 69–72. [Google Scholar]
- Alsonosi, A.; Hariri, S.; Kajsík, M.; Oriešková, M.; Hanulík, V.; Röderová, M.; Petrželová, J.; Kollárová, H.; Drahovská, H.; Forsythe, S.; et al. The speciation and genotyping of Cronobacter isolates from hospitalised patients. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1979–1988. [Google Scholar] [CrossRef] [Green Version]
- Yong, W.; Guo, B.; Shi, X.; Cheng, T.; Chen, M.; Jiang, X.; Ye, Y.; Wang, J.; Xie, G.; Ding, J. An investigation of an acute gastroenteritis outbreak: Cronobacter sakazakii, a potential cause of food-borne illness. Front. Microbiol. 2018, 9, 2549. [Google Scholar] [CrossRef]
- Patrick, M.E.; Mahon, B.E.; Greene, S.A.; Rounds, J.; Cronquist, A.; Wymore, K.; Boothe, E.; Lathrop, S.; Palmer, A.; Bowen, A. Incidence of Cronobacter spp. infections, United States, 2003–2009. Emerg. Infect. Dis. 2014, 20, 1520–1523. [Google Scholar] [CrossRef]
- Lepuschitz, S.; Ruppitsch, W.; Pekard-Amenitsch, S.; Forsythe, S.J.; Cormican, M.; Mach, R.L.; Piérard, D.; Allerberger, F.; EUCRONI Study Group. Multicenter Study of Cronobacter sakazakii infections in humans, Europe, 2017. Emerg. Infect. Dis. 2019, 25, 515–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strysko, J.; Cope, J.R.; Martin, H.; Tarr, C.; Hise, K.; Collier, S.; Bowen, A. Food safety and invasive Cronobacter infections during early infancy, 1961–2018. Emerg. Infect. Dis. 2020, 26, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.; Gopinath, G.R.; Eshwar, A.; Srikumar, S.; Nguyen, S.; Gangiredla, J.; Patel, I.R.; Finkelstein, S.B.; Negrete, F.; Woo, J.; et al. The secretion of toxins and other exoproteins of Cronobacter: Role in virulence, adaption, and persistence. Microorganisms 2020, 8, 229. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.; Sonbol, H.; Hariri, S.; Desai, P.; MacClelland, M.; Forsythe, S.J. Diversity of the Cronobacter genus as revealed by multilocus sequence typing. J. Clin. Microbiol. 2012, 50, 3031–3039. [Google Scholar] [CrossRef] [Green Version]
- Himelright, I.; Harris, E.; Lorch, V.; Anderson, M.; Jones, T.; Craig, A.; Kuehnert, M.; Forster, T.; Arduino, M.; Jensen, B.; et al. Enterobacter sakazakii infections associated with the use of powdered infant formula–Tennessee, 2001. Morb. Mortal. Wkly. Rep. 2002, 51, 297–300. [Google Scholar]
- Enterobacter sakazakii and Other Micro-Organisms in Powdered Infant Formula: Meeting Report; Microbiol. Risk Assessment Series; FAO/WHO: Rome, Italy, 2004. Available online: http://www.fao.org/3/a-y5502e.pdf (accessed on 10 November 2021).
- Jason, J. Prevention of invasive Cronobacter infections in young infants fed powdered infant formulas. Pediatrics 2012, 130, e1076–e1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, M.; Fouladkhah, A. Outbreak history, biofilm formation, and preventive measures for control of Cronobacter sakazakii in infant formula and infant care settings. Microorganisms 2019, 7, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandhai, M.C.; Heuvelink, A.E.; Reij, M.W.; Beumer, R.R.; Dijk, R.; van Tilburg, J.J.H.C.; van Schothorst, M.; Gorris, L.G.M. A study into the occurrence of Cronobacter spp. in The Netherlands between 2001 and 2005. Food Control 2010, 21, 1127–1136. [Google Scholar] [CrossRef]
- Lou, X.; Si, G.; Yu, H.; Qi, J.; Liu, T.; Fang, Z. Possible reservoir and routes of transmission of Cronobacter (Enterobacter sakazakii) via wheat flour. Food Control 2014, 43, 258–262. [Google Scholar] [CrossRef]
- Sani, N.A.; Odeyemi, O.A. Occurrence and prevalence of Cronobacter spp. in plant and animal derived food sources: A systematic review and meta-analysis. SpringerPlus 2015, 4, 545. [Google Scholar] [CrossRef] [Green Version]
- Berthold-Pluta, A.; Garbowska, M.; Stefańska, I.; Pluta, A. Microbiological quality of selected ready-to-eat leaf vegetables, sprouts and non-pasteurized fresh fruit-vegetable juices including the presence of Cronobacter spp. Food Microbiol. 2017, 65, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Brandão, M.L.L.; Umeda, N.S.; Jackson, E.; Forsythe, S.J.; de Filippis, I. Isolation, molecular and phenotypic characterization, and antibiotic susceptibility of Cronobacter spp. from Brazilian retail foods. Food Microbiol. 2017, 63, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Moravkova, M.; Verbikova, V.; Huvarova, V.; Babak, V.; Cahlikova, H.; Karpiskova, R.; Kralik, P. Occurrence of Cronobacter spp. in ready-to-eat vegetable products, frozen vegetables, and sprouts examined using cultivation and real-time PCR methods. J. Food Sci. 2018, 83, 3054–3058. [Google Scholar] [CrossRef] [PubMed]
- Vasconcellos, L.; Carvalho, C.T.; Tavares, R.O.; de Mello Medeiros, V.; de Oliveira Rosas, C.; Silva, J.N.; Dos Reis Lopes, S.M.; Forsythe, S.J.; Brandão, M.L.L. Isolation, molecular and phenotypic characterization of Cronobacter spp. in ready-to-eat salads and foods from Japanese cuisine commercialized in Brazil. Food Res. Int. 2018, 107, 353–359. [Google Scholar] [CrossRef]
- Hayman, M.; Edelson-Mammel, S.; Carter, P.; Chen, Y.; Metz, M.; Sheehan, J.F.; Tall, B.D.; Thompson, C.J.; Smoot, L.A. Prevalence of Cronobacter spp. and Salmonella in milk powder manufacturing facilities in the United States. J. Food Prot. 2020, 10, 4315. [Google Scholar] [CrossRef]
- Gan, X.; Li, M.; Yan, S.; Wang, X.; Wang, W.; Li, F. Genomic Landscape and phenotypic Assessment of Cronobacter sakazakii isolated from raw material, environment, and production facilities in powdered infant formula factories in China. Front. Microbiol. 2021, 12, 686189. [Google Scholar] [CrossRef]
- Pava-Ripoll, M.; Pearson, R.E.; Miller, A.K.; Ziobro, G.C. Prevalence and relative risk of Cronobacter spp., Salmonella spp., and Listeria monocytogenes associated with the body surfaces and guts of individual filth flies. Appl. Environ. Microbiol. 2012, 78, 7891–7902. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Gopinath, G.R.; Chase, H.R.; Gangiredla, J.; Eshwar, A.; Patel, I.; Addy, N.; Ewing, L.; Beaubrun, J.J.G.; Negrete, F.; et al. Draft genomes of Cronobacter sakazakii strains isolated from dried spices bring unique insights into the epidemiology of plant-associated strains. Stand. Genom. Sci. 2018, 13, 35. [Google Scholar] [CrossRef]
- Jang, H.; Chase, H.R.; Gangiredla, J.; Grim, C.J.; Patel, I.R.; Kothary, M.H.; Jackson, S.A.; Mammel, M.K.; Carter, L.; Negrete, F.; et al. Analysis of the molecular diversity among Cronobacter species isolated from filth flies using targeted PCR, pan genomic DNA microarray, and whole genome sequencing analyses. Front. Microbiol. 2020, 11, 561204. [Google Scholar] [CrossRef]
- Ueda, S. Occurrence of Cronobacter spp. in dried foods, fresh vegetables, and soil. Biocontrol Sci. 2017, 22, 55–59. [Google Scholar] [CrossRef] [Green Version]
- Fei, P.; Jiang, Y.; Gong, S.; Li, R.; Jiang, Y.; Yuan, X.; Wang, Z.; Kang, H.; Ali, M.A. Occurrence, genotyping, and antibiotic susceptibility of Cronobacter spp. in drinking water and food samples from Northeast China. J. Food Prot. 2018, 23, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Ling, N.; Li, C.; Zhang, J.; Wu, Q.; Zeng, H.; He, W.; Ye, Y.; Wang, J.; Ding, Y.; Chen, M.; et al. Prevalence and molecular and antimicrobial characteristics of Cronobacter spp. isolated from raw vegetables in China. Front. Microbiol. 2018, 9, 1149. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Iversen, C.; Gontia, I.; Stephan, R.; Hofmann, A.; Hartmann, A.; Jha, B.; Eberl, L.; Riedel, K.; Lehner, A. Evidence for a plant associated natural habitat for Cronobacter spp. Res. Microbiol. 2009, 160, 608–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, H.R.; Eberl, L.; Stephan, R.; Jeong, H.; Lee, C.; Finkelstein, S.; Negrete, F.; Gangiredla, J.; Patel, I.; Tall, B.D.; et al. Draft genome sequence of Cronobacter sakazakii GP1999, sequence type 145, an epiphytic isolate obtained from the tomato’s rhizoplane/rhizosphere continuum. Genome Announc. 2017, 5, e00723-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, H.R.; Gopinath, G.R.; Eshwar, A.K.; Stoller, A.; Fricker-Feer, C.; Gangiredla, J.; Patel, I.R.; Cinar, H.N.; Jeong, H.; Lee, C.; et al. Comparative genomic characterization of the highly persistent and potentially virulent Cronobacter sakazakii ST83, CC65 strain H322 and other ST83 strains. Front. Microbiol. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Gopinath, G.R.; Chase, H.R.; Gangiredla, J.; Eschwar, A.; Jang, H.; Patel, I.; Negrete, F.; Finkelstein, S.; Park, E.; Chung, T.; et al. Genomic characterization of malonate positive Cronobacter sakazakii serotype O:2, sequence type 64 strains, isolated from clinical, food, and environment samples. Gut Pathog. 2018, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Addy, N.; Ewing, L.; Beaubrun, J.J.-G.; Lee, Y.; Woo, J.; Negrete, F.; Finkelstein, S.; Tall, B.D.; Lehner, A.; et al. Whole-Genome sequences of Cronobacter sakazakii isolates obtained from foods of plant origin and dried-food manufacturing environments. Genome Announc. 2018, 6, e00223-18. [Google Scholar] [CrossRef] [Green Version]
- Eida, A.A.; Bougouffa, S.; L’Haridon, F.; Alam, I.; Weisskopf, L.; Bajic, V.B.; Saad, M.M.; Hirt, H. Genome insights of the plant-growth promoting bacterium Cronobacter muytjensii Z38 with volatile-mediated antagonistic activity against Phytophthora infestans. Front. Microbiol 2020, 11, 369. [Google Scholar] [CrossRef] [Green Version]
- Grim, C.J.; Kotewicz, M.L.; Power, K.A.; Gopinath, G.; Franco, A.A.; Jarvis, K.G.; Yan, Q.Q.; Jackson, S.A.; Sathyamoorthy, V.; Hu, L.; et al. Pan-genome analysis of the emerging foodborne pathogen Cronobacter spp. suggests a species-level bidirectional divergence driven by niche adaptation. BMC Genomics. 2013, 14, 366. [Google Scholar] [CrossRef] [Green Version]
- Stoop, B.; Lehner, A.; Iversen, C.; Fanning, S.; Stephan, R. Development and evaluation of rpoB based PCR systems to differentiate the six proposed species within the genus Cronobacter. Int. J. Food Microbiol. 2009, 136, 165–168. [Google Scholar] [CrossRef] [Green Version]
- Lehner, A.; Fricker-Feer, C.; Stephan, R. Identification of the recently described Cronobacter condimenti by an rpoB gene-based PCR system. J. Med. Microbiol. 2012, 61, 1034–1035. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.; Lindsey, L.; Grim, C.J.; Sathyamoorthy, V.; Jarvis, K.G.; Gopinath, G.; Lee, C.; Sadowski, J.A.; Trach, L.; Pava-Ripoll, M.; et al. Multiplex PCR assay targeting a diguanylate cyclase-encoding gene, cgcA, to differentiate species within the genus Cronobacter. Appl. Environ. Microbiol. 2013, 79, 734–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q.Q.; Jarvis, K.G.; Chase, H.R.; Hébert, K.; Trach, L.H.; Lee, C.; Sadowski, J.; Lee, B.; Hwang, S.; Sathyamoorthy, V.; et al. A proposed harmonized LPS molecular-subtyping scheme for Cronobacter species. Food Microbiol. 2015, 50, 38–43. [Google Scholar] [CrossRef]
- Franco, A.A.; Hu, L.; Grim, C.J.; Gopinath, G.; Sathyamoorthy, V.; Jarvis, K.G.; Lee, C.; Sadowski, J.; Kim, J.; Kothary, M.H.; et al. Characterization of putative virulence genes on the related RepFIB plasmids harbored by Cronobacter spp. Appl. Environ. Microbiol. 2011, 77, 3255–3267. [Google Scholar] [CrossRef] [Green Version]
- Kucerova, E.; Clifton, S.W.; Xia, X.Q.; Long, F.; Porwollik, S.; Fulton, L.; Fronick, C.; Minx, P.; Kyung, K.; Warren, W.; et al. Genome sequence of Cronobacter sakazakii BAA-894 and comparative genomic hybridization analysis with other Cronobacter species. PLoS ONE 2010, 5, e9556. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 8, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangiredla, J.; Rand, H.; Benisatto, D.; Payne, J.; Strittmatter, C.; Sanders, J.; Wolfgang, W.J.; Libuit, K.; Herrick, J.B.; Prarat, M.; et al. GalaxyTrakr: A distributed analysis tool for public health whole genome sequence data accessible to non-bioinformaticians. BMC Genom. 2021, 22, 114. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Shi, L.; Liang, Q.; Zhan, Z.; Feng, J.; Zhao, Y.; Chen, Y.; Huang, M.; Tong, Y.; Wu, W.; Chen, W.; et al. Co-occurrence of 3 different resistance plasmids in a multi-drug resistant Cronobacter sakazakii isolate causing neonatal infections. Virulence 2018, 9, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16-21. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [Green Version]
- Tall, B.D.; Gangiredla, J.; Gopinath, G.R.; Yan, Q.; Chase, H.R.; Lee, B.; Hwang, S.; Trach, L.; Park, E.; Yoo, Y.; et al. Development of a custom-designed, pan genomic DNA microarray to characterize strain-level diversity among Cronobacter spp. Front. Pediatr. 2015, 3, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.A.; Patel, I.R.; Barnaba, T.; LeClerc, J.E.; Cebula, T.A. Investigating the global diversity of Escherichia coli using a multi-genome DNA microarray platform with novel gene prediction strategies. BMC Genom. 2011, 12, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affymetrix. Expression Analysis Technical Manual, with Specific Protocols for Use with the Hybridization, Wash, and Stain Kit. 2014. Available online: http://www.affymetrix.com/support/downloads/manuals/expression_analysis_technical_manual.pdf (accessed on 10 November 2021).
- Tall, B.D.; Gangiredla, J.; Grim, C.J.; Patel, I.R.; Jackson, S.A.; Mammel, M.K.; Gopinath, G.R. Use of a pan–genomic DNA microarray in determination of the phylogenetic relatedness among Cronobacter spp. and its use as a data mining tool to understand Cronobacter biology. Microarrays 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Fehr, A.; Eshwar, A.K.; Neuhauss, S.C.; Ruetten, M.; Lehner, A.; Vaughan, L. Evaluation of the Zebrafish as a model to study the pathogenesis of the opportunistic pathogen Cronobacter turicensis. Emerg. Microbes Infect. 2015, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Eshwar, A.K.; Tall, B.D.; Gangiredla, J.; Gopinath, G.R.; Patel, I.R.; Neuhauss, S.C.F.; Stephan, R.; Lehner, A. Linking genomo- and pathotype: Exploiting the Zebrafish embryo model to investigate the divergent virulence potential among Cronobacter spp. PLoS ONE 2016, 11, e0158428. [Google Scholar] [CrossRef]
- Moine, D.; Kassam, M.; Baert, L.; Tang, Y.; Barretto, C.; Ngom Bru, C.; Klijn, A.; Descombes, P. Fully closed genome sequences of five type strains of the genus Cronobacter and one Cronobacter sakazakii strain. Genome Announc. 2016, 4, e00142-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q.; Power, K.A.; Cooney, S.; Fox, E.; Gopinath, G.R.; Grim, C.J.; Tall, B.D.; McCusker, M.P.; Fanning, S. Complete genome sequence and phenotype microarray analysis of Cronobacter sakazakii SP291: A persistent isolate cultured from a powdered infant formula production facility. Front. Microbiol. 2013, 4, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaradat, Z.W.; Ababneh, Q.O.; Saadoun, I.M.; Samara, N.A.; Rashdan, A.M. Isolation of Cronobacter spp. (formerly Enterobacter sakazakii) from infant food, herbs and environmental samples and the subsequent identification and confirmation of the isolates using biochemical, chromogenic assays, PCR and 16S rRNA sequencing. BMC Microbiol. 2009, 9, 225. [Google Scholar] [CrossRef] [Green Version]
- Chon, J.W.; Song, K.Y.; Kim, S.Y.; Hyeon, J.Y.; Seo, K.H. Isolation and characterization of Cronobacter from desiccated foods in Korea. J. Food Sci. 2012, 77, M354–M358. [Google Scholar] [CrossRef]
- Bentley, S.D.; Parkhill, J. Genomic perspectives on the evolution and spread of bacterial pathogens. Proc. Biol. Sci. 2015, 282, 20150488. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.; Hachler, H.; Stephan, R.; Lehner, A. Presence of AmpC Beta-Lactamases, CSA-1, CSA-2, CMA-1, and CMA-2 Conferring an Unusual Resistance Phenotype in Cronobacter sakazakii and Cronobacter malonaticus. Microb. Drug Resist. 2014, 20, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EMA/CVMP/CHMP/231573/2016; Updated Advice on the Use of Colistin Products in Animals within the European Union: Development of Resistance and Possible Impact on Human and Animal Health. EMA: Amsterdam, The Netherlands, 2016.
- Liu, B.-T.; Song, F.-J.; Zou, M.; Hao, Z.-H.; Shan, H. Emergence of colistin resistance gene mcr-1 in Cronobacter sakazakii producing NDM-9 and in Escherichia coli from the same animal. Antimicrob. Agents Chemother. 2017, 61, e01444-16. [Google Scholar] [CrossRef] [Green Version]
- Arcilla, M.S.; van Hattem, J.M.; Matamoros, S.; Melles, D.C.; Penders, J.; de Jong, M.D.; Schultsz, C. COMBAT Consortium. Dissemination of the mcr-1 colistin resistance gene. Lancet Infect. Dis. 2016, 16, 147–149. [Google Scholar] [CrossRef] [Green Version]
- Falgenhauer, L.; Waezsada, S.E.; Yao, Y.; Imirzalioglu, C.; Kasbohrer, A.; Roesler, U.; Michael, G.B.; Schwarz, S.; Werner, G.; Kreienbrock, L.; et al. RESET consortium. Colistin resistance gene mcr-1 in extended-spectrum beta-lactamase-producing and carbapenemase-producing Gram-negative bacteria in Germany. Lancet Infect. Dis. 2016, 16, 282–283. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Ohnishi, M.; Kawanishi, M.; Akiba, M.; Kuroda, M. Investigation of a plasmid genome database for colistin-resistance gene mcr-1. Lancet Infect. Dis. 2016, 16, 284–285. [Google Scholar] [CrossRef] [Green Version]
- Stoesser, N.; Mathers, A.J.; Moore, C.E.; Day, N.P.; Cook, D.W. Colistin resistance gene mcr-1 and pHNSHP45 plasmid in human isolates of Escherichia coli and Klebsiella pneumoniae. Lancet Infect. Dis. 2016, 16, 285–286. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, L.; Feng, Y.; He, D.; Wang, C.; Zong, Z. Potential mobilization of mcr-10 by an integrative mobile element via site-specific recombination in Cronobacter sakazakii. Antimicrob. Agents Chemother. 2021, 65, e01717-20. [Google Scholar] [CrossRef] [PubMed]
- Hölzel, C.S.; Tetens, J.L.; Schwaiger, K. Unraveling the role of vegetables in spreading antimicrobial-resistant bacteria: A need for quantitative risk assessment. Foodborne Pathog. Dis. 2018, 15, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Fortier, L.C.; Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 2013, 4, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Zhang, J.; Li, C.; Xie, T.; Ling, N.; Wu, Q.; Ye, Y. The driving force of prophages and CRISPR-Cas system in the evolution of Cronobacter sakazakii. Sci. Rep. 2017, 7, 40206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Choi, Y.; Shin, H.; Lee, J.; Ryu, S. Complete genome sequence of Cronobacter sakazakii temperate bacteriophage phiES15. J. Virol. 2012, 86, 7713–7714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.D.; Park, J.H. Complete genome of temperate phage ENT39118 from Cronobacter sakazakii. J. Virol. 2012, 9, 5400–5401. [Google Scholar] [CrossRef] [Green Version]
- Negrete, F.J.; Ko, K.; Jang, H.; Hoffmann, M.; Lehner, A.; Stephan, R.; Fanning, S.; Tall, B.D.; Gopinath, G.R. Complete genome sequences and genomic characterization of five plasmids harbored by environmentally persistent Cronobacter sakazakii strains ST83 H322 and ST64 GK1025B obtained from powdered infant formula manufacturing facilities. Gut Pathog. 2022, 14, 23. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Shaker, R.R.; Osaili, T.M.; Abu Al-Hasan, A.S.; Ayyash, M.M.; Forsythe, S.J. Effect of desiccation, starvation, heat, and cold stresses on the thermal resistance of Enterobacter sakazakii in rehydrated infant milk formula. J. Food Sci. 2008, 73, M354–M359. [Google Scholar] [CrossRef] [PubMed]
- Osaili, T.; Forsythe, S. Desiccation resistance and persistence of Cronobacter species in infant formula. Int. J. Food Microbiol. 2009, 136, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Lehner, A.; Tall, B.D.; Fanning, S.; Shabarinath, S. Cronobacter spp.–Opportunistic foodborne pathogens: An update on evolution, osmotic adaptation and pathogenesis. Curr. Clin. Microbiol. Rep. 2018, 5, 97–105. [Google Scholar] [CrossRef]
- Yan, Q.Q.; Wang, J.; Gangiredla, J.; Cao, Y.; Martins, M.; Gopinath, G.R.; Stephan, R.; Lampel, K.; Tall, B.; Fanning, S. Comparative genotypic and phenotypic analysis of Cronobacter species cultured from four powdered infant formula production facilities: Indication of patho-adaptation along the food chain. Appl. Environ. Microbiol. 2015, 13, 4388–4403. [Google Scholar] [CrossRef] [Green Version]
- Negrete, F.; Gangiredla, J.; Jang, H.; Chase, H.R.; Woo, J.; Lee, Y.; Patel, I.; Finkelstein, S.B.; Tall, B.D.; Gopinath, G.R. Prevalence and distribution of efflux pump complexes genes in Cronobacter sakazakii using whole genome and pan-genomic datasets. Curr. Opin. Food Sci. 2019, 30, 32–42. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Strain | Source | Country | Genome Size (kb) | G + C Contents (%) | No. of CDSs | ST a, CC b | Serotype f | NCBI Accession No. |
|---|---|---|---|---|---|---|---|---|
| MOD1_LR607 | Corn flour | USA | 4577 | 56.8 | 4399 | 13, CC13 | CsakO:4 | PVCO00000000 |
| MOD1_LR626 | Rice flour | USA | 4547 | 56.8 | 4345 | 1, CC1 | CsakO:1 | PVCR00000000 |
| MOD1_LR627 | Corn flour | USA | 4405 | 56.8 | 4204 | 1, CC1 | CsakO:1 | PVCM00000000 |
| MOD1_LR636 | Sodium caseinate | USA | 4420 | 57 | 4177 | 148, CC16 | CsakO:1 | PVCN00000000 |
| MOD1_LR637 | Sodium caseinate | USA | 4421 | 57 | 4176 | 148, CC16 | CsakO:1 | PVCS00000000 |
| MOD1_LR639 | Sodium caseinate | USA | 4557 | 56.7 | 4365 | 1, CC1 | CsakO:1 | PVCP00000000 |
| MOD1_LR641 | Sodium caseinate | USA | 4534 | 56.7 | 4297 | 21, CC21 | CsakO:1 | PVCQ00000000 |
| MOD1_LR643 | Soy flour | USA | 4500 | 56.9 | 4284 | 73, CC73 | CsakO:7 | PVDI00000000 |
| MOD1_LR646 | Soy flour | USA | 4522 | 56.8 | 4307 | 73, CC73 | CsakO:7 | PVDJ00000000 |
| MOD1_LR688 | Org c flour | USA | 4403 | 57 | 4169 | 1, CC1 | CsakO:1 | PVDF00000000 |
| MOD1_LR702 | Powdered infant formula | USA | 4648 | 56.7 | 4449 | 21, CC21 | CsakO:1 | PVDG00000000 |
| MOD1_LR703 | Flour | USA | 4428 | 57.1 | 4180 | 22 | CsakO:2 | PVCT00000000 |
| MOD1_LR704 | Flour | USA | 4404 | 57 | 4188 | 1, CC1 | CsakO:1 | QHGX00000000 |
| MOD1_LR705 | Org. soy powder | USA | 4481 | 56.9 | 4325 | 4, CC4 | CsakO:2 | QHGW00000000 |
| MOD1_LR706 | Org. soy powder | USA | 4488 | 56.9 | 4304 | 4, CC4 | CsakO:2 | PVCU00000000 |
| MOD1_LR711 | Org. soy powder | USA | 4545 | 56.7 | 4371 | 1, CC1 | CsakO:1 | PVCV00000000 |
| MOD1_LR712 | Org. soy powder | USA | 4472 | 56.9 | 4318 | ND d | CsakO:2 | PVCW00000000 |
| MOD1_LR713 | Environment | USA | 4465 | 57 | 4266 | 83, CC83 | CsakO:7 | PTOX00000000 |
| MOD1_LR714 | Org. soy powder | USA | 4553 | 56.7 | 4360 | 1, CC1 | CsakO:1 | PVCX00000000 |
| MOD1_LR721 | Org. soy powder | USA | 4401 | 57 | 4144 | 12 | CsakO:4 | PVCY00000000 |
| MOD1_LR723 | Org. flour powder | USA | 4471 | 56.9 | 4266 | 1, CC1 | CsakO:1 | PVCZ00000000 |
| MOD1_LR728 | Org. soy | USA | 4550 | 56.7 | 4403 | 1, CC1 | CsakO:1 | PVMX00000000 |
| MOD1_LR735 | Whey protein | USA | 4675 | 56.6 | 4474 | 1, CC1 | CsakO:1 | PVDH00000000 |
| MOD1_LR736 | Whey protein | USA | 4679 | 56.7 | 4475 | 1, CC1 | CsakO:1 | PVDA00000000 |
| MOD1_LR745 | Unknown food powder | USA | 4598 | 56.9 | 4395 | 4, CC4 | CsakO:2 | PVMY00000000 |
| MOD1_LR747 | Seasoning powder blend e | USA | 4675 | 56.7 | 4540 | 13, CC13 | CsakO:2 | PVDB00000000 |
| MOD1_LR751 | Org. soy powder | USA | 4436 | 56.8 | 4251 | 4, CC4 | CsakO:3 | PVDC00000000 |
| MOD1_LR755 | Org. soy powder | USA | 4557 | 56.7 | 4364 | 1, CC1 | CsakO:1 | PVDD00000000 |
| MOD1_LR756 | Org. flour powder | USA | 4577 | 56.8 | 4405 | 13, CC13 | CsakO:2 | PVDE00000000 |
| MOD1_Jor100 | Semolina | Jordan | 4362 | 57 | 4181 | 643 | CsakO:2 | NITS00000000 |
| MOD1_Jor175 | Spice | Jordan | 4364 | 56.9 | 4121 | 1, CC1 | CsakO:1 | NITO00000000 |
| MOD1_Jor204 | Liquorice | Jordan | 4352 | 57.1 | 4097 | 223 | CsakO:7 | QHGV00000000 |
| MOD1_KW9 | Sorghum | Republic of Korea | 4610 | 56.8 | 4405 | 143, CC143 | ND f | NITF00000000 |
| Gene | Sequence Name | Subclass | Strain |
|---|---|---|---|
| blaCSA-1 | Class C beta-lactamase CSA-1 | Cephalothin | Jor20, Jor22 (2 strains) b |
| blaCSA-2 | Class C beta-lactamase CSA-2 | Cephalothin | LR631, LR632, LR705, LR706, LR712, LR715, LR745, LR751, Jor96, Jor148, Jor154, Jor178 (12 strains) c |
| blaCSA | CSA family class C beta-lactamase | Cephalothin | LR607, LR626, LR627, LR634, LR635, LR636, LR637, LR639, LR640, LR641, LR643, LR646, LR654, LR688, LR702, LR703, LR704, LR707, LR708, LR711, LR713, LR714, LR721, LR722, LR723, LR728, LR733, LR735, LR736, LR747, LR752, LR753, LR755, LR756, LR757, KW1, KW3, KW4, KW9, KW11, KW13, KW18, 1-15, 3-21, 5-17G, 5-20G, 5-21G, O21-13, O21-16, O23mB, O26-1, O26-4, WNTSBCO4, 16MP2184, 16MP2185, 760029, 777122, 788569, AS-13, AS-15, AS-2, AS-4, Jor44, Jor93, Jor100, Jor103, Jor109, Jor146, Jor151, Jor172, Jor173, Jor175, Jor183, Jor204 (74 strains) d |
| mcr-9.1 | Phosphoethanolamine-lipid A transferase mrc-9.1 | Colistin | LR735, LR736, WNTSBOCO4 |
| Code | Value | % Avg a | Description |
|---|---|---|---|
| A | 1 | 0.0 | RNA processing and modification |
| B | 0 | 0.0 | Chromatin structure and dynamics |
| C | 211 | 5.5 | Energy production and conversion |
| D | 45 | 1.2 | Cell cycle control, Cell division, chromosome partitioning |
| E | 293 | 7.6 | Amino acid transport and metabolism |
| F | 100 | 2.6 | Nucleotide transport and metabolism |
| G | 328 | 8.5 | Carbohydrate transport and metabolism |
| H | 140 | 3.6 | Coenzyme transport and metabolism |
| I | 79 | 2.0 | Lipid transport and metabolism |
| J | 180 | 4.7 | Translation, ribosomal structure and biogenesis |
| K | 298 | 7.7 | Transcription |
| L | 183 | 4.7 | Replication, recombination and repair |
| M | 259 | 6.7 | Cell wall/membrane biogenesis |
| N | 67 | 1.7 | Cell motility |
| O | 151 | 3.9 | Posttranslational modification, protein turnover, chaperones |
| P | 247 | 6.4 | Inorganic ion transport and metabolism |
| Q | 45 | 1.2 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 0 | 0.0 | General function prediction only |
| S | 1056 | 27.3 | Function unknown |
| T | 158 | 4.1 | Signal transduction mechanisms |
| U | 66 | 1.7 | Intracellular trafficking and secretion |
| V | 48 | 1.3 | Defense mechanisms |
| - | 0 | 0.0 | Not in COGs |
| Total No. of C. sak Isolates | pESA3/ pCTU1 (incFIB) | pESA2 /pCTU2(incFII) | pCTU3 /H1 | No. of Isolates with the Indicated Plasmidotype (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cpa | T6SS | FHA | Iron Acquisition | |||||||||||
| cpaa | Δcpa | Int L | vgrG | R end | Int R | ΔT6SS | fhaB | ΔFHA | eitA | iucC | ||||
| 92 | 92 (100) | 5 (6) | 20 (30) | 91 (99) | 0 (0) | 91 (99) | 50 (54) | 56 (60) | 29 (31) | 0 (0) | 19 (21) | 66 (72) | 92 (100) | 92 (100) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, H.; Eshwar, A.; Lehner, A.; Gangiredla, J.; Patel, I.R.; Beaubrun, J.J.-G.; Chase, H.R.; Negrete, F.; Finkelstein, S.; Weinstein, L.M.; et al. Characterization of Cronobacter sakazakii Strains Originating from Plant-Origin Foods Using Comparative Genomic Analyses and Zebrafish Infectivity Studies. Microorganisms 2022, 10, 1396. https://doi.org/10.3390/microorganisms10071396
Jang H, Eshwar A, Lehner A, Gangiredla J, Patel IR, Beaubrun JJ-G, Chase HR, Negrete F, Finkelstein S, Weinstein LM, et al. Characterization of Cronobacter sakazakii Strains Originating from Plant-Origin Foods Using Comparative Genomic Analyses and Zebrafish Infectivity Studies. Microorganisms. 2022; 10(7):1396. https://doi.org/10.3390/microorganisms10071396
Chicago/Turabian StyleJang, Hyein, Athmanya Eshwar, Angelika Lehner, Jayanthi Gangiredla, Isha R. Patel, Junia Jean-Gilles Beaubrun, Hannah R. Chase, Flavia Negrete, Samantha Finkelstein, Leah M. Weinstein, and et al. 2022. "Characterization of Cronobacter sakazakii Strains Originating from Plant-Origin Foods Using Comparative Genomic Analyses and Zebrafish Infectivity Studies" Microorganisms 10, no. 7: 1396. https://doi.org/10.3390/microorganisms10071396