
Phylogenomic Analysis of Salmonella enterica subsp. enterica Serovar Bovismorbificans from Clinical and Food Samples Using Whole Genome Wide Core Genes and kmer Binning Methods to Identify Two Distinct Polyphyletic Genome Pathotypes

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Genomic DNA Preparation
2.3. Whole Genome Sequencing (WGS), Assembly, and Annotation
2.4. Identification of Whole-Genome Core Genes and High-Resolution Phylogenomic Analysis
2.5. Data Mining of a DNA Tiling Microarray Database for Genomic Comparisons with Legacy Strain Collections
2.6. Characterization of Multi-Locus Sequence Typing (MLST) and Antimicrobial Resistance Gene (AMR) Patterns
2.7. Identification of Salmonella Pathogenicity Islands (SPIs), Plasmids, and Prophage Genomic Regions and Virulence Factors
3. Results and Discussion
3.1. Genome Sequencing, Assembly and MLST Typing
3.2. Data Mining of DNA Tiling Digital Hybridization Profiles Identified Distinct Genomic Backbone Differences
3.3. Development of a Workflow for Core Genome Analysis Based on Complete Genomes from 150 S. enterica Serovars
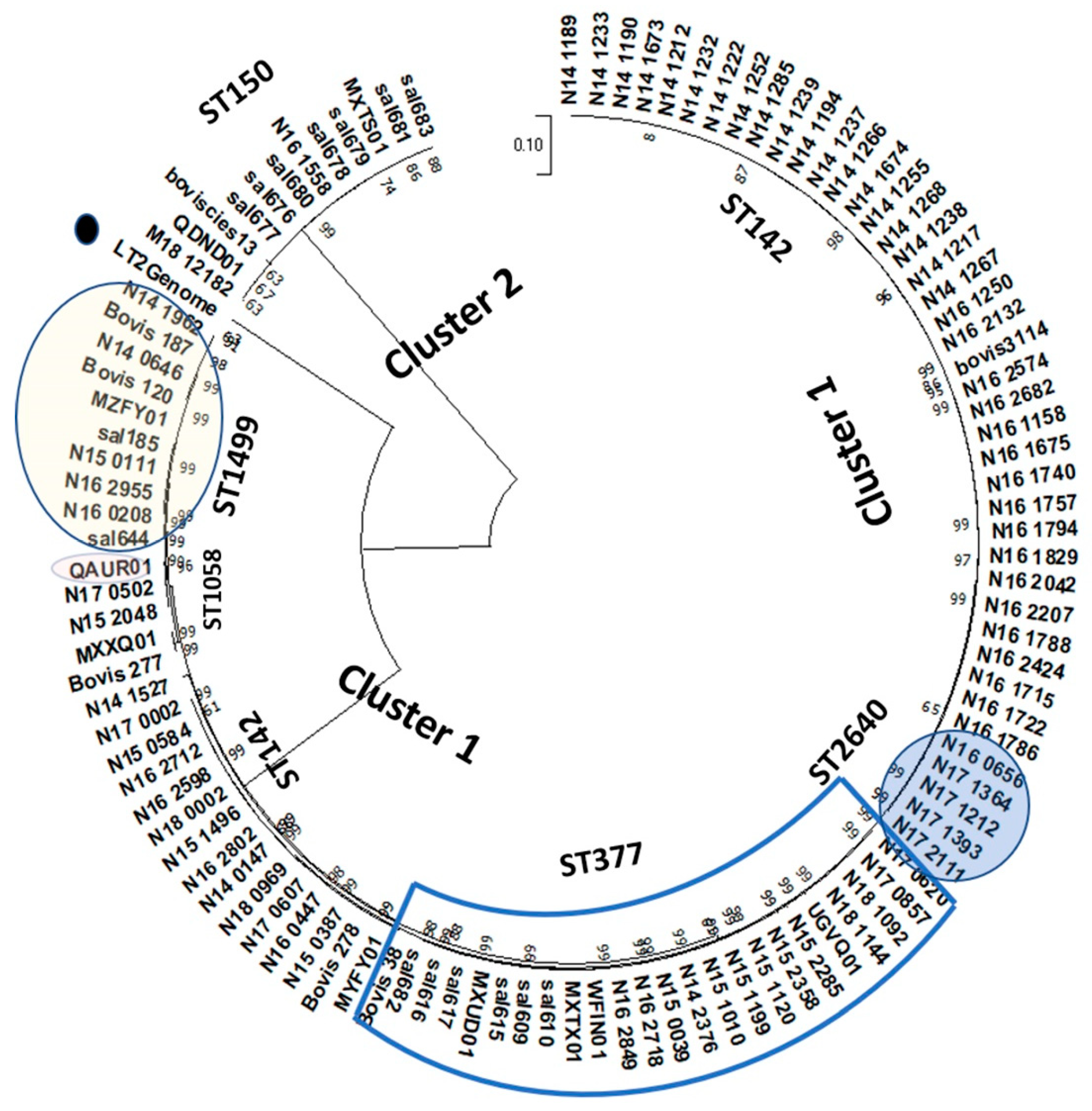
3.4. Genome Pathotyping of S. Bovismorbificans Strains Using Wg-Core Genes and k-Mer-Binning Methods
3.5. Plasmids, Phage, AMR, and Virulence Profiling of S. Bovismorbificans Genome Types
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Havelaar, A.H.; Kirk, M.D.; Torgerson, P.R.; Gibb, H.J.; Hald, T.; Lake, R.J.; Praet, N.; Bellinger, D.C.; de Silva, N.R.; Gargouri, N.; et al. World Health Organization Global Estimates and Regional Comparisons of the Burden of Foodborne Disease in 2010. PLoS Med. 2015, 12, e1001923. [Google Scholar] [CrossRef]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States–major pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef]
- Nguyen, D.T.; Kanki, M.; Nguyen, P.D.; Le, H.T.; Ngo, P.T.; Tran, D.N.; Le, N.H.; Dang, C.V.; Kawai, T.; Kawahara, R.; et al. Prevalence, antibiotic resistance, and extended-spectrum and AmpC β-lactamase productivity of Salmonella isolates from raw meat and seafood samples in Ho Chi Minh City, Vietnam. Int. J. Food Microbiol. 2016, 236, 115–122. [Google Scholar] [CrossRef]
- Pönkä, A.; Andersson, Y.; Siitonen, A.; de Yong, B.; Jahkola, M.; Haikala, O.; Kuhmonen, A.; Pakkala, P. Salmonella in alfalfa sprouts. Lancet 1995, 345, 462–463. [Google Scholar] [CrossRef]
- Puohiniemi, R.; Heiskanen, T.; Siitonen, A. Molecular epidemiology of two international sprout-borne Salmonella outbreaks. J. Clin. Microbiol. 1997, 35, 2487–2491. [Google Scholar] [CrossRef]
- Rimhanen-Finne, R.; Niskanen, R.T.; Lienemann, T.; Johansson, T.; Sjöman, M.; Korhonen, T.; Guedes, S.; Kuronen, H.; Virtanen, M.J.; Mä Kinen, J.; et al. Nationwide outbreak of Salmonella Bovismorbificans associated with sprouted alfalfa seeds in Finland, 2009. Zoonoses Public Health 2011, 58, 589–596. [Google Scholar] [CrossRef]
- Blaylock, M.; Blackwell, R.; Merid, S.; Jackson, S.; Kotewicz, M.; Gopinath, G.; Ayers, S.L.; Abbott, J.; Sabo, J.; Ewing, L.; et al. Comparison of Salmonella enterica serovar Bovismorbificans 2011 hummus outbreak strains with non-outbreak strains. Food Microbiol. 2015, 46, 627–634. [Google Scholar] [CrossRef]
- Knoblauch, A.M.; Bratschi, M.W.; Zuske, M.K.; Althaus, D.; Stephan, R.; Hächler, H.; Baumgartner, A.; Prager, R.; Rabsch, W.; Altpeter, E.; et al. Cross-border outbreak of Salmonella enterica ssp. enterica serovar Bovismorbificans: Multiple approaches for an outbreak investigation in Germany and Switzerland. Swiss Med. Wkly. 2015, 145, w14182. [Google Scholar] [CrossRef]
- Bronowski, C.; Fookes, M.C.; Gilderthorp, R.; Ashelford, K.E.; Harris, S.R.; Phiri, A.; Hall, N.; Gordon, M.A.; Wain, J.; Hart, C.A.; et al. Genomic characterisation of invasive non-typhoidal Salmonella enterica Subspecies enterica Serovar Bovismorbificans isolates from Malawi. PLoS Negl. Trop. Dis. 2013, 7, e2557. [Google Scholar] [CrossRef]
- Brandwagt, D.; van den Wijngaard, C.; Tulen, A.D.; Mulder, A.C.; Hofhuis, A.; Jacobs, R.; Heck, M.; Verbruggen, A.; van den Kerkhof, H.; Slegers-Fitz-James, I.; et al. Outbreak of Salmonella Bovismorbificans associated with the consumption of uncooked ham products, the Netherlands, 2016 to 2017. Eurosurveillance 2018, 23, 17-00335. [Google Scholar] [CrossRef]
- Gopinath, G.; Jean-Gilles Beaubrun, J.; Grim, C.; Hanes, D. Draft Genome Sequences of Nine Salmonella enterica Serovar Bovismorbificans Isolates from Various Sources. Genome Announc. 2014, 2, e01249-13. [Google Scholar] [CrossRef]
- CDC. Multistate Outbreak of Salmonella serotype Bovismorbificans Infections Associated with Hummus and Tahini d United States. 2011. Available online: www.cdc.gov/mmwr/preview/mmwrhtml/mm6146a3.htm (accessed on 6 April 2022).
- Centers for Disease Control and Prevention (CDC). Multistate outbreak of Salmonella serotype Bovismorbificans infections associated with hummus and tahini—United States, 2011. MMWR Morb. Mortal. Wkly. Rep. 2012, 61, 944–947. [Google Scholar]
- Gopinath, G.; Jean-Gilles Beaubrun, J.; Grim, C.; Blaylock, M.; Blackwell, R.; Merid, S.; Diallo, A.; Hanes, D. Whole-Genome Sequences of Six Salmonella enterica Serovar Bovismorbificans Isolates Associated with a 2011 Multistate Hummus-Borne Outbreak. Genome Announc. 2014, 2, e01239-13. [Google Scholar] [CrossRef]
- Nógrády, N.; Imre, A.; Kostyák, A.; Tóth, A.; Nagy, B. Molecular and pathogenic characterization of Salmonella enterica serovar Bovismorbificans strains of animal, environmental, food, and human origin in Hungary. Foodborne Pathog. Dis. 2010, 7, 507–513. [Google Scholar] [CrossRef]
- Cheng, R.A.; Eade, C.R.; Wiedmann, M. Embracing Diversity: Differences in Virulence Mechanisms, Disease Severity, and Host Adaptations Contribute to the Success of Nontyphoidal Salmonella as a Foodborne Pathogen. Front. Microbiol. 2019, 10, 1368. [Google Scholar] [CrossRef]
- Achtman, M.; Wain, J.; Weill, F.X.; Nair, S.; Zhou, Z.; Sangal, V.; Krauland, M.G.; Hale, J.L.; Harbottle, H.; Uesbeck, A.; et al. Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog. 2012, 8, e1002776, Erratum in: PLoS Pathog. 2020, 16, e1009040. [Google Scholar] [CrossRef]
- Den Bakker, H.C.; Moreno Switt, A.I.; Govoni, G.; Cummings, C.A.; Ranieri, M.L.; Degoricija, L.; Hoelzer, K.; Rodriguez-Rivera, L.D.; Brown, S.; Bolchacova, E.; et al. Genome sequencing reveals diversification of virulence factor content and possible host adaptation in distinct subpopulations of Salmonella enterica. BMC Genom. 2011, 12, 425. [Google Scholar] [CrossRef]
- Timme, R.E.; Pettengill, J.B.; Allard, M.W.; Strain, E.; Barrangou, R.; Wehnes, C.; VanKessel, J.S.; Karns, J.S.; Musser, S.M.; Brown, E.W. Phylogenetic diversity of the enteric pathogen Salmonella enterica subsp. enterica inferred from genome-wide reference-free SNP characters. Genome Biol. Evol. 2013, 5, 2109–2123. [Google Scholar] [CrossRef]
- Cao, G.; Meng, J.; Strain, E.; Stones, R.; Pettengill, J.; Zhao, S.; McDermott, P.; Brown, E.; Allard, M. Phylogenetics and differentiation of Salmonella Newport lineages by whole genome sequencing. PLoS ONE 2013, 8, e55687. [Google Scholar] [CrossRef]
- Mastrorilli, E.; Petrin, S.; Orsini, M.; Longo, A.; Cozza, D.; Luzzi, I.; Ricci, A.; Barco, L.; Losasso, C. Comparative genomic analysis reveals high intra-serovar plasticity within Salmonella Napoli isolated in 2005–2017. BMC Genom. 2020, 21, 202. [Google Scholar] [CrossRef]
- Shariat, N.W.; Timme, R.E.; Walters, A.T. Phylogeny of Salmonella enterica subspecies arizonae by whole-genome sequencing reveals high incidence of polyphyly and low phase 1 H antigen variability. Microb. Genom. 2021, 7, 000522. [Google Scholar] [CrossRef]
- Grimont, P.A.D.; Weill, F.X. Antigenic Formulae of the Salmonella Serovars, 9th ed.; World Health Organization Collaborating Centre for Reference and Research on Salmonella, Institut Pasteur: Geneva, Switzerland, 2007. [Google Scholar]
- Jean-Gilles Beaubrun, J.; Cheng, C.; Chen, K.; Ewing, L.; Wang, H.; Agpaoa, M.C.; Huang, M.J.; Dickey, E.; Du, J.M.; Williams-Hill, D.M.; et al. The use of a PCR-based method for identification of Salmonella enterica serotypes Newport, Typhimurium, Javiana and Saint Paul from various food matrices. Food Microbiol. 2012, 31, 199–209. [Google Scholar] [CrossRef]
- Zhang, S.; Yin, Y.; Jones, M.B.; Zhang, Z.; Deatherage Kaiser, B.L.; Dinsmore, B.A.; Fitzgerald, C.; Fields, P.I.; Deng, X. Salmonella serotype determination utilizing high-throughput genome sequencing data. J. Clin. Microbiol. 2015, 53, 1685–1692. [Google Scholar] [CrossRef]
- Gangiredla, J.; Rand, H.; Benisatto, D.; Payne, J.; Strittmatter, C.; Sanders, J.; Wolfgang, W.J.; Libuit, K.; Herrick, J.B.; Prarat, M.; et al. GalaxyTrakr: A distributed analysis tool for public health whole genome sequence data accessible to non-bioinformaticians. BMC Genom. 2021, 22, 114. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Haft, D.H.; DiCuccio, M.; Badretdin, A.; Brover, V.; Chetvernin, V.; O’Neill, K.; Li, W.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; et al. RefSeq: An update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018, 46, D851–D860. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.W.; Strain, E.; Melka, D.; Bunning, K.; Musser, S.M.; Brown, E.W.; Timme, R. Practical Value of Food Pathogen Traceability through Building a Whole-Genome Sequencing Network and Database. J. Clin. Microbiol. 2016, 54, 1975–1983. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef]
- Saitou, M.; Nei, N. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Jackson, S.A.; Patel, I.R.; Barnaba, T.; LeClerc, J.E.; Cebula, T.A. Investigating the global diversity of Escherichia coli using a multi-genome DNA microarray platform with novel gene prediction strategies. BMC Genom. 2011, 12, 349. [Google Scholar] [CrossRef]
- Li, B.; Jackson, S.; Gangiredla, J.; Wang, W.; Liua, H.; Tall, B.D.; Jean-Gilles Beaubrun, J.; Jay-Russell, M.; Vellidis, G.; Elkins, C.A. Genomic Evidence Reveals Numerous Re-Introduction Events of Salmonella enterica serovar Newport in Irrigation Ponds in Suwannee Watershed. Appl. Environ. Microbiol. 2015, 81, 8243–8253. [Google Scholar] [CrossRef]
- Affymetrix. Expression Analysis Technical Manual, with Specific Protocols for Use with the Hybridization, Wash, and Stain Kit. 2014. Available online: http://www.affymetrix.com/support/downloads/manuals/expression_analysis_technical_manual.pdf (accessed on 6 April 2022).
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Achtman, M.; Hale, J.; Murphy, R.A.; Boyd, E.F.; Porwollik, S. Population structures in the SARA and SARB reference collections of Salmonella enterica according to MLST, MLEE and microarray hybridization. Infect. Genet. Evol. 2013, 16, 314–325. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.H.; McDermott, P.F.; et al. Validating the AMRFinder Tool and Resistance Gene Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of Isolates. Antimicrob. Agents Chemother. 2019, 63, e00483-19. [Google Scholar] [CrossRef]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Maiden, M.C.; Jansen van Rensburg, M.J.; Bray, J.E.; Earle, S.G.; Ford, S.A.; Jolley, K.A.; McCarthy, N.D. MLST revisited: The gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 2013, 11, 728–736. [Google Scholar] [CrossRef]
- Achtman, M.; Zhou, Z.; Alikhan, N.F.; Tyne, W.; Parkhill, J.; Cormican, M.; Chiou, C.S.; Torpdahl, M.; Litrup, E.; Prendergast, D.M.; et al. Genomic diversity of Salmonella enterica-The UoWUCC 10K genomes project. Wellcome Open Res. 2020, 5, 22. [Google Scholar] [CrossRef]
- Worley, J.; Meng, J.; Allard, M.W.; Brown, E.W.; Timme, R.E. Salmonella enterica Phylogeny Based on Whole-Genome Sequencing Reveals Two New Clades and Novel Patterns of Horizontally Acquired Genetic Elements. mBio 2018, 9, e02303-18. [Google Scholar] [CrossRef]
- Deng, X.; den Bakker, H.C.; Hendriksen, R.S. Genomic Epidemiology: Whole-Genome-Sequencing-Powered Surveillance and Outbreak Investigation of Foodborne Bacterial Pathogens. Annu. Rev. Food Sci. Technol. 2016, 7, 353–374. [Google Scholar] [CrossRef]
- Yoshida, C.E.; Kruczkiewicz, P.; Laing, C.R.; Lingohr, E.J.; Gannon, V.P.; Nash, J.H.; Taboada, E.N. The Salmonella In Silico Typing Resource (SISTR): An Open Web-Accessible Tool for Rapidly Typing and Subtyping Draft Salmonella Genome Assemblies. PLoS ONE 2016, 11, e0147101. [Google Scholar] [CrossRef]
- Pearce, M.E.; Alikhan, N.F.; Dallman, T.J.; Zhou, Z.; Grant, K.; Maiden, M.C.J. Comparative analysis of core genome MLST and SNP typing within a European Salmonella serovar Enteritidis outbreak. Int. J. Food Microbiol. 2018, 274, 1–11. [Google Scholar] [CrossRef]
- Alikhan, N.F.; Zhou, Z.; Sergeant, M.J.; Achtman, M. A genomic overview of the population structure of Salmonella. PLoS Genet. 2018, 14, e1007261. [Google Scholar] [CrossRef]
- Timme, R.E.; Strain, E.; Baugher, J.D.; Davis, S.; Gonzalez-Escalona, N.; Sanchez Leon, M.; Allard, M.W.; Brown, E.W.; Tallent, S.; Rand, H. Phylogenomic Pipeline Validation for Foodborne Pathogen Disease Surveillance. J. Clin. Microbiol. 2019, 57, e01816-18. [Google Scholar] [CrossRef]
- Barretto, C.; Rincón, C.; Portmann, A.C.; Ngom-Bru, C. Whole Genome Sequencing Applied to Pathogen Source Tracking in Food Industry: Key Considerations for Robust Bioinformatics Data Analysis and Reliable Results Interpretation. Genes 2021, 12, 275. [Google Scholar] [CrossRef]
- Brown, E.; Dessai, U.; McGarry, S.; Gerner-Smidt, P. Use of Whole-Genome Sequencing for Food Safety and Public Health in the United States. Foodborne Pathog Dis. 2019, 16, 441–450. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Kaas, R.S.; Seyfarth, A.M.; Agersø, Y.; Lund, O.; Larsen, M.V.; Aarestrup, F.M. Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. Antimicrob. Chemother. 2013, 68, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Branchu, P.; Bawn, M.; Kingsley, R.A. Genome Variation and Molecular Epidemiology of Salmonella enterica Serovar Typhimurium Pathovariants. Infect. Immun. 2018, 86, e00079-18. [Google Scholar] [CrossRef]
- Cavaco, L.M.; Hasman, H.; Xia, S.; Aarestrup, F.M. qnrD, a novel gene conferring transferable quinolone resistance in Salmonella enterica Serovar Kentucky and Bovismorbificans strains of human origin. Antimicrob. Agents Chemother. 2009, 53, 603–608. [Google Scholar] [CrossRef]
- Finley, R.; Reid-Smith, R.; Ribble, C.; Popa, M.; Vandermeer, M.; Aramini, J. The occurrence and anti-microbial susceptibility of Salmonellae isolated from commercially available pig ear pet treats. Zoonoses Public Health. 2008, 55, 455–461. [Google Scholar]
- Jajere, S.M. A review of Salmonella enterica with particular focus on the pathogenicity and virulence factors, host specificity and antimicrobial resistance including multidrug resistance. Vet. World 2019, 12, 504–521. [Google Scholar] [CrossRef]
- Hanna, L.F.; Matthews, T.D.; Dinsdale, E.A.; Hasty, D.; Edwards, R.A. Characterization of the ELPhiS prophage from Salmonella enterica serovar Enteritidis strain LK5. Appl. Environ. Microbiol. 2012, 78, 1785–1793. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Source | Country | Year | Genome Size (kb) | No. of CDSs | ST a | NCBI Biosample | NCBI Accession No. | Reference |
|---|---|---|---|---|---|---|---|---|---|
| M18_12182 | Environment | USA | 2018 | 4679 | 4399 | 150 | SAMN12657258 | WSCV00000000 | This study |
| Bovis_38 | Beef | Canada | 1989 | 4579 | 4321 | 142 | SAMN12657217 | WSCW00000000 | This study |
| Bovis_120 | Seafood, squid | Canada | missing | 4706 | 4474 | 1499 | SAMN12657218 | WSCX00000000 | This study |
| Bovis_187 | Seafood, shrimp | Canada | 1989 | 4739 | 4498 | 1499 | SAMN12657204 | WSCY00000000 | This study |
| Bovis_277 | Clinical | Canada | 1984 | 4832 | 4638 | 1499 | SAMN12657210 | WSCZ00000000 | This study |
| Bovis_278 | Clinical | Canada | 1984 | 4738 | 4498 | 142 | SAMN12657232 | WSDA00000000 | This study |
| N14_0147 | Clinical | Switzerland | 2014 | 4690 | 4471 | 142 | SAMN12657231 | WSDB00000000 | This study |
| N14_0646 | Clinical | Switzerland | 2014 | 4702 | 4449 | 1499 | SAMN12657228 | WSDC00000000 | This study |
| N14_1189 | Clinical | Switzerland | 2014 | 4790 | 4575 | 142 | SAMN12657257 | WSDD00000000 | This study |
| N14_1190 | Clinical | Switzerland | 2014 | 4784 | 4566 | 142 | SAMN12657229 | WSDE00000000 | This study |
| N14_1194 | Clinical | Switzerland | 2014 | 4789 | 4580 | 142 | SAMN12657219 | WSDF00000000 | This study |
| N14_1212 | Clinical | Switzerland | 2014 | 4790 | 4577 | 142 | SAMN12657254 | WSDG00000000 | This study |
| N14_1217 | Clinical | Switzerland | 2014 | 4854 | 4688 | 142 | SAMN12657212 | WSDH00000000 | This study |
| N14_1222 | Clinical | Switzerland | 2014 | 4791 | 4577 | 142 | SAMN12657214 | WSDI00000000 | This study |
| N14_1232 | Clinical | Switzerland | 2014 | 4778 | 4560 | 142 | SAMN12657205 | WSDJ00000000 | This study |
| N14_1233 | Clinical | Switzerland | 2014 | 4790 | 4568 | 142 | SAMN12657209 | WSDK00000000 | This study |
| N14_1237 | Clinical | Switzerland | 2014 | 4781 | 4565 | 142 | SAMN12657230 | WSDL00000000 | This study |
| N14_1238 | Clinical | Switzerland | 2014 | 4781 | 4563 | 142 | SAMN12657235 | WSDM00000000 | This study |
| N14_1239 | Clinical | Switzerland | 2014 | 4778 | 4562 | 142 | SAMN12657165 | WSDN00000000 | This study |
| N14_1252 | Clinical | Switzerland | 2014 | 4781 | 4562 | 142 | SAMN12657181 | WSDO00000000 | This study |
| N14_1255 | Clinical | Switzerland | 2014 | 4786 | 4576 | 142 | SAMN12657154 | WSDP00000000 | This study |
| N14_1266 | Clinical | Switzerland | 2014 | 4779 | 4561 | 142 | SAMN12657194 | WSDQ00000000 | This study |
| N14_1267 | Clinical | Switzerland | 2014 | 4775 | 4602 | 142 | SAMN12657197 | WSDR00000000 | This study |
| N14_1268 | Clinical | Switzerland | 2014 | 4783 | 4567 | 142 | SAMN12657192 | WSDS00000000 | This study |
| N14_1285 | Clinical | Switzerland | 2014 | 4780 | 4561 | 142 | SAMN12657190 | WSDT00000000 | This study |
| N14_1527 | Clinical | Switzerland | 2014 | 4813 | 4590 | 1499 | SAMN12657150 | WSDU00000000 | This study |
| N14_1673 | Clinical | Switzerland | 2014 | 4819 | 4609 | 142 | SAMN12657188 | WSDV00000000 | This study |
| N14_1674 | Clinical, blood | Switzerland | 2014 | 4783 | 4571 | 142 | SAMN12657156 | WSDW00000000 | This study |
| N14_1962 | Clinical | Switzerland | 2014 | 4715 | 4468 | 1499 | SAMN12657164 | WSDX00000000 | This study |
| N14_2376 | Food, onion | Switzerland | 2014 | 4723 | 4486 | 377 | SAMN12657147 | WSDY00000000 | This study |
| N15_0039 | Food, grain | Switzerland | 2015 | 4726 | 4493 | 377 | SAMN12657185 | WSDZ00000000 | This study |
| N15_0111 | Clinical | Switzerland | 2015 | 5024 | 4860 | 1499 | SAMN12657157 | WSEA00000000 | This study |
| N15_0387 | Clinical | Switzerland | 2015 | 4643 | 4404 | 142 | SAMN12657198 | WSEB00000000 | This study |
| N15_0584 | Clinical | Switzerland | 2015 | 4670 | 4468 | 142 | SAMN12657182 | WSEC00000000 | This study |
| N15_1010 | Food | Switzerland | 2015 | 4725 | 4483 | 377 | SAMN12657196 | WSED00000000 | This study |
| N15_1120 | Food | Switzerland | 2015 | 4724 | 4486 | 377 | SAMN12657193 | WSEE00000000 | This study |
| N15_1199 | Food | Switzerland | 2015 | 4720 | 4485 | 377 | SAMN12657191 | WSEF00000000 | This study |
| N15_1496 | Clinical | Switzerland | 2015 | 4738 | 4530 | 142 | SAMN12657189 | WSEG00000000 | This study |
| N15_2048 | Clinical | Switzerland | 2015 | 4847 | 4671 | 1499 | SAMN12657202 | WSEH00000000 | This study |
| N15_2285 | Clinical | Switzerland | 2015 | 4806 | 4598 | 377 | SAMN12657199 | WSEI00000000 | This study |
| N15_2358 | Clinical | Switzerland | 2015 | 4893 | 4724 | 377 | SAMN12657233 | WSEJ00000000 | This study |
| N16_0208 | Clinical | Switzerland | 2016 | 4798 | 4571 | 1499 | SAMN12657155 | WSBH00000000 | This study |
| N16_0447 | Clinical, urine | Switzerland | 2016 | 4636 | 4399 | 142 | SAMN12657160 | WSBI00000000 | This study |
| N16_0656 | Clinical | Switzerland | 2016 | 4838 | 4628 | 2640 | SAMN12657256 | WSBJ00000000 | This study |
| N16_1158 | Clinical | Switzerland | 2016 | 4781 | 4566 | 142 | SAMN12657253 | WSBK00000000 | This study |
| N16_1250 | Clinical | Switzerland | 2016 | 4788 | 4571 | 142 | SAMN12657211 | WSBL00000000 | This study |
| N16_1558 | Clinical | Switzerland | 2016 | 4571 | 4290 | 150 | SAMN12657220 | WSBM00000000 | This study |
| N16_1675 | Clinical | Switzerland | 2016 | 4782 | 4573 | 142 | SAMN12657207 | WSBN00000000 | This study |
| N16_1715 | Clinical | Switzerland | 2016 | 4784 | 4570 | 142 | SAMN12657200 | WSBO00000000 | This study |
| N16_1722 | Clinical | Switzerland | 2016 | 4785 | 4569 | 142 | SAMN12657206 | WSBP00000000 | This study |
| N16_1740 | Clinical | Switzerland | 2016 | 4778 | 4556 | 142 | SAMN12657234 | WSBQ00000000 | This study |
| N16_1757 | Clinical | Switzerland | 2016 | 4784 | 4568 | 142 | SAMN12657203 | WSBR00000000 | This study |
| N16_1786 | Clinical | Switzerland | 2016 | 4781 | 4567 | 142 | SAMN12657259 | WSBS00000000 | This study |
| N16_1788 | Clinical | Switzerland | 2016 | 4784 | 4584 | 142 | SAMN12657260 | WSBT00000000 | This study |
| N16_1794 | Clinical | Switzerland | 2016 | 4794 | 4573 | 142 | SAMN12657221 | WSBU00000000 | This study |
| N16_1829 | Clinical | Switzerland | 2016 | 4785 | 4571 | 142 | SAMN12657255 | WSBV00000000 | This study |
| N16_2042 | Clinical | Switzerland | 2016 | 4778 | 4561 | 142 | SAMN12657208 | WSBW00000000 | This study |
| N16_2132 | Clinical | Switzerland | 2016 | 4787 | 4563 | 142 | SAMN12657216 | WSBX00000000 | This study |
| N16_2207 | Animal, cat | Switzerland | 2016 | 4778 | 4573 | 142 | SAMN12657146 | WSBY00000000 | This study |
| N16_2424 | Clinical | Switzerland | 2016 | 4775 | 4556 | 142 | SAMN12657163 | WSBZ00000000 | This study |
| N16_2574 | Clinical | Switzerland | 2016 | 4947 | 4754 | 142 | SAMN12657148 | WSCA00000000 | This study |
| N16_2598 | Clinical | Switzerland | 2016 | 4706 | 4481 | 142 | SAMN12657161 | WSCB00000000 | This study |
| N16_2682 | Clinical | Switzerland | 2016 | 4777 | 4574 | 142 | SAMN12657159 | WSCC00000000 | This study |
| N16_2712 | Clinical | Switzerland | 2016 | 4703 | 4489 | 142 | SAMN12657184 | WSCD00000000 | This study |
| N16_2718 | Clinical | Switzerland | 2016 | 4723 | 4482 | 377 | SAMN12657144 | WSCE00000000 | This study |
| N16_2802 | Clinical | Switzerland | 2016 | 4724 | 4508 | 142 | SAMN12657195 | WSCF00000000 | This study |
| N16_2849 | Clinical | Switzerland | 2016 | 4725 | 4489 | 377 | SAMN12657142 | WSCG00000000 | This study |
| N16_2955 | Clinical | Switzerland | 2016 | 4811 | 4599 | 1499 | SAMN12657187 | WSCH00000000 | This study |
| N17_0002 | Clinical | Switzerland | 2017 | 4706 | 4492 | 142 | SAMN12657149 | WSCI00000000 | This study |
| N17_0502 | Clinical | Switzerland | 2017 | 4873 | 4696 | 1499 | SAMN12657152 | WSCJ00000000 | This study |
| N17_0607 | Clinical | Switzerland | 2017 | 4640 | 4401 | 142 | SAMN12657145 | WSCK00000000 | This study |
| N17_0620 | Food | Switzerland | 2017 | 4731 | 4493 | 377 | SAMN12657151 | WSCL00000000 | This study |
| N17_0857 | Feed | Switzerland | 2017 | 4724 | 4507 | 377 | SAMN12657158 | WSCM00000000 | This study |
| N17_1212 | Clinical | Switzerland | 2017 | 4773 | 4576 | 2640 | SAMN12657162 | WSCN00000000 | This study |
| N17_1364 | Clinical | Switzerland | 2017 | 4869 | 4680 | 2640 | SAMN12657186 | WSCO00000000 | This study |
| N17_1393 | Clinical | Switzerland | 2017 | 4779 | 4564 | 2640 | SAMN12657153 | WSCP00000000 | This study |
| N17_2111 | Clinical | Switzerland | 2017 | 4775 | 4561 | 2640 | SAMN12657143 | WSCQ00000000 | This study |
| N18_0002 | Clinical | Switzerland | 2018 | 4805 | 4605 | 142 | SAMN12657183 | WSCR00000000 | This study |
| N18_0969 | Clinical | Switzerland | 2018 | 4640 | 4402 | 142 | SAMN12657213 | WSCS00000000 | This study |
| N18_1092 | Clinical | Switzerland | 2018 | 4728 | 4492 | 377 | SAMN12657215 | WSCT00000000 | This study |
| N18_1144 | Clinical | Switzerland | 2018 | 4722 | 4544 | 377 | SAMN12657201 | WSCU00000000 | This study |
| Sal609 b | Clinical | USA | 2011 | 4896 | 4925 | 377 | SAMN02422699 | AZKW00000000 | [11] |
| Sal610 | Clinical | USA | 2011 | 4857 | 4870 | 377 | SAMN02422700 | AZKX00000000 | [11] |
| Sal615 | Clinical | USA | 2011 | 4845 | 4891 | 377 | SAMN02422701 | AZKY00000000 | [11] |
| Sal616 | Food | USA | 2011 | 4865 | 4893 | 377 | SAMN02422702 | AZKZ00000000 | [11] |
| Sal617 | Food | USA | 2011 | 4872 | 4887 | 377 | SAMN02422703 | AZLA00000000 | [11] |
| Sal644 | Clinical | USA | 2001 | 4769 | 4769 | 1499 | SAMN02422688 | AZLC00000000 | [11] |
| Sal676 | Clinical | USA | 2012 | 4569 | 4476 | 150 | SAMN02422698 | AZKV00000000 | [11] |
| Sal677 | Clinical | USA | 2012 | 4663 | 4614 | 150 | SAMN02422693 | AZKR00000000 | [14] |
| Sal678 | Clinical | USA | 2012 | 4579 | 4629 | 150 | SAMN02422694 | AZKS00000000 | [14] |
| Sal679 | Clinical | USA | 2012 | 4567 | 4471 | 150 | SAMN02422695 | AZKT00000000 | [14] |
| Sal680 | Clinical | USA | 2012 | 4596 | 4506 | 150 | SAMN02422696 | AZKU00000000 | [14] |
| Sal681 | Clinical | USA | 2012 | 4575 | 4492 | 150 | SAMN02422697 | AZLB00000000 | [14] |
| Sal682 | Clinical | USA | 2012 | 4926 | 4944 | 377 | SAMN02422690 | AZKQ00000000 | [11] |
| Sal683 | Clinical | USA | 2012 | 4574 | 4586 | 150 | SAMN02422689 | AZKP00000000 | [14] |
| bovis3114 b | Clinical | Malawi | 1997 | 4680 | 4599 | 142 | SAMEA3138815 | HF969015 | [9] |
| bovispt13 | Unknown | Unknown | Unknown | NA c | NA | Unknown d | SAMN01081634 | SRS347148 e | NCBI SRA |
| boviscies13 | Water | Mexico | 2013 | NA | NA | 150 | SAMN02335370 | SRS476367 f | NCBI SRA |
| pSal610 | Clinical | USA | 2011 | 93.8 | 111 | 377 | SAMN02422700 | CP076746 | This study |
| Strain | Gene | Subclass | Sequence Name/Description |
|---|---|---|---|
| N14_0147 | aph(3″)-Ib | Streptomycin | Aminoglycoside O-phosphotransferase APH(3″)-Ib |
| sul2 | Sulfonamide | Sulfonamide-resistant dihydropteroate synthase Sul2 | |
| aph(6)-Id | Streptomycin | Aminoglycoside O-phosphotransferase APH(6)-Id | |
| N15_0111 | blaCTX-M-55 | Cephalosporin | Class A extended-spectrum beta-lactamase CTX-M-55 |
| qnrS1 | Quinolone | Quinolone resistance pentapeptide repeat protein QnrS1 | |
| tet(A) | Tetracycline | Tetracycline efflux MFS transporter Tet(A) | |
| blaTEM-1 | Beta-lactam | Class A broad-spectrum beta-lactamase TEM-1 | |
| tet(M) | Tetracycline | Tetracycline resistance ribosomal protection protein Tet(M) | |
| aac(3)-IId | Gentamicin | Aminoglycoside N-acetyltransferase AAC(3)-IId | |
| aph(3′)-IIa | Kanamycin | Aminoglycoside O-phosphotransferase APH(3′)-IIa | |
| ble | Bleomycin | Bleomycin binding protein BLMT | |
| bleO | Bleomycin | Bleomycin binding protein | |
| sul3 | Sulfonamide | Sulfonamide-resistant dihydropteroate synthase Sul3 | |
| aadA2 | Streptomycin | ANT(3″)-Ia family aminoglycoside Nucleotidyltransferase AadA2 | |
| cmlA1 | Chloramphenicol | Chloramphenicol efflux MFS transporter CmlA1 | |
| aadA1 | Streptomycin | ANT(3″)-Ia family aminoglycoside Nucleotidyltransferase AadA1 | |
| qacL | Quaternary ammonium b | Quaternary ammonium compound efflux SMR transporter QacL | |
| N16_0208 | aph(6)-Id | Streptomycin | Aminoglycoside O-phosphotransferase APH(6)-Id |
| sul2 | Sulfonamide | Sulfonamide-resistant dihydropteroate synthase Sul2 | |
| aph(3″)-Ib | Streptomycin | Aminoglycoside O-phosphotransferase APH(3″)-Ib | |
| tet(A) | Tetracycline | Tetracycline efflux MFS transporter Tet(A) | |
| N16_2574 | dfrA1 | Trimethoprim | Trimethoprim-resistant dihydrofolate reductase DfrA1 |
| blaTEM-1 | Beta-lactam | Class A broad-spectrum beta-lactamase TEM-1 | |
| sul2 | Sulfonamide | Sulfonamide-resistant dihydropteroate synthase Sul2 | |
| blaCTX-M-1 | Cephalosporin | Class A extended-spectrum beta-lactamase CTX-M-1 | |
| N16_2598 | tet(A) | Tetracycline | Tetracycline efflux MFS transporter Tet(A) |
| N16_2955 | aph(3″)-Ib | Streptomycin | Aminoglycoside O-phosphotransferase APH(3″)-Ib |
| aph(6)-Id | Streptomycin | Aminoglycoside O-phosphotransferase APH(6)-Id | |
| tet(A) | Tetracycline | Tetracycline efflux MFS transporter Tet(A) | |
| sul2 | Sulfonamide | Sulfonamide-resistant dihydropteroate synthase Sul2 | |
| N17_0502 | floR | Chloramphenicol/florfenicol | Chloramphenicol/florfenicol efflux MFS transporter FloR |
| qnrB19 | Quinolone | Quinolone resistance pentapeptide repeat protein QnrB19 | |
| sul2 | Sulfonamide | Sulfonamide-resistant dihydropteroate synthase Sul2 | |
| aph(3″)-Ib | Streptomycin | Aminoglycoside O-phosphotransferase APH(3″)-Ib | |
| aph(6)-Id | Streptomycin | Aminoglycoside O-phosphotransferase APH(6)-Id | |
| tet(A) | Tetracycline | Tetracycline efflux MFS transporter Tet(A) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gopinath, G.R.; Jang, H.; Beaubrun, J.J.-G.; Gangiredla, J.; Mammel, M.K.; Müller, A.; Tamber, S.; Patel, I.R.; Ewing, L.; Weinstein, L.M.; et al. Phylogenomic Analysis of Salmonella enterica subsp. enterica Serovar Bovismorbificans from Clinical and Food Samples Using Whole Genome Wide Core Genes and kmer Binning Methods to Identify Two Distinct Polyphyletic Genome Pathotypes. Microorganisms 2022, 10, 1199. https://doi.org/10.3390/microorganisms10061199
Gopinath GR, Jang H, Beaubrun JJ-G, Gangiredla J, Mammel MK, Müller A, Tamber S, Patel IR, Ewing L, Weinstein LM, et al. Phylogenomic Analysis of Salmonella enterica subsp. enterica Serovar Bovismorbificans from Clinical and Food Samples Using Whole Genome Wide Core Genes and kmer Binning Methods to Identify Two Distinct Polyphyletic Genome Pathotypes. Microorganisms. 2022; 10(6):1199. https://doi.org/10.3390/microorganisms10061199
Chicago/Turabian StyleGopinath, Gopal R., Hyein Jang, Junia Jean-Gilles Beaubrun, Jayanthi Gangiredla, Mark K. Mammel, Andrea Müller, Sandeep Tamber, Isha R. Patel, Laura Ewing, Leah M. Weinstein, and et al. 2022. "Phylogenomic Analysis of Salmonella enterica subsp. enterica Serovar Bovismorbificans from Clinical and Food Samples Using Whole Genome Wide Core Genes and kmer Binning Methods to Identify Two Distinct Polyphyletic Genome Pathotypes" Microorganisms 10, no. 6: 1199. https://doi.org/10.3390/microorganisms10061199