
Gastro-Intestinal Microbiota in Equines and Its Role in Health and Disease: The Black Box Opens
Abstract
:1. Characteristics of Horse GIT Microbiota along the Gastrointestinal Tract and Its Temporal Evolution during Life
1.1. The “Common Core Microbiota” of the Adult Healthy Horse: A Myth or a Reality?
1.2. Evolution of the Horse Microbiota with Age
2. Horse GIT Microbiota Changes According to Abiotic Factors
2.1. Impact of Diet Composition
2.2. Transportation
2.3. High Performance
2.4. Heat Stress Conditions
3. Horse GIT Microbiota and Digestive Disorders: What Do we Know?
3.1. Equine Gastric Ulcer Syndrom (EGUS)
3.2. Colitis and Diarrhea
3.3. Colic
3.4. Free Fecal Water
3.5. Parasitism
4. Systemic Disorders: Laminitis, Equine Metabolic Syndrome (EMS) and Obesity and Their Relationships with the Horse GIT Microbiome
5. Discussion: The Microbiota of Horses, a Crucial “Organ” Largely Associated with Health and Diseases in Horses
6. Conclusions and Future Directions: An Exciting Field of Research to Support Digestion, Performance and Health of Horses, but Still a Lot of Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Julliand, V.; Grimm, P. HORSE SPECIES SYMPOSIUM: The microbiome of the horse hindgut: History and current knowledge. J. Anim. Sci. 2016, 94, 2262–2274. [Google Scholar] [CrossRef]
- Kauter, A.; Epping, L.; Semmler, T.; Antao, E.-M.; Kannapin, D.; Stoeckle, S.D.; Gehlen, H.; Lübke-Becker, A.; Günther, S.; Wieler, L.H.; et al. The gut microbiome of horses: Current research on equine enteral microbiota and future perspectives. Anim. Microbiome 2019, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.C.; Silva, G.; Ramos, R.V.; Staempfli, H.R.; Arroyo, L.G.; Kim, P.; Weese, J.S. Characterization and comparison of the bacterial microbiota in different gastrointestinal tract compartments in horses. Vet. J. 2015, 205, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Risely, A. Applying the core microbiome to understand host-microbe systems. J. Anim. Ecol. 2020, 89, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Dougal, K.; Harris, P.A.; Edwards, A.; Pachebat, J.A.; Blackmore, T.M.; Worgan, H.J.; Newbold, C.J. A comparison of the microbiome and the metabolome of different regions of the equine hindgut. FEMS Microbiol. Ecol. 2012, 82, 642–652. [Google Scholar] [CrossRef]
- Schoster, A.; Arroyo, L.G.; Staempfli, H.R.; Weese, J.S. Comparison of microbial populations in the small intestine, large intestine and feces of healthy horses using terminal restriction fragment length polymorphism. BMC Res. Notes 2013, 6, 91. [Google Scholar] [CrossRef]
- Su, S.; Zhao, Y.; Liu, Z.; Liu, G.; Du, M.; Wu, J.; Bai, D.; Li, B.; Bou, G.; Zhang, X.; et al. Characterization and comparison of the bacterial microbiota in different gastrointestinal tract compartments of Mongolian horses. Microbiologyopen 2020, 9, 1085–1101. [Google Scholar] [CrossRef]
- De Fombelle, A.; Varloud, M.; Goachet, A.-G.; Jacotot, E.; Philippeau, C.; Drogoul, C.; Julliand, V. Characterization of the microbial and biochemical profile of the different segments of the digestive tract in horses given two distinct diets. Anim. Sci. 2003, 77, 293–304. [Google Scholar] [CrossRef]
- Ericsson, A.C.; Johnson, P.J.; Lopes, M.A.; Perry, S.C.; Lanter, H.R. A Microbiological Map of the Healthy Equine Gastrointestinal Tract. PLoS ONE 2016, 11, e0166523. [Google Scholar] [CrossRef]
- Perkins, G.A.; den Bakker, H.C.; Burton, A.J.; Erb, H.N.; McDonough, S.P.; McDonough, P.L.; Parker, J.; Rosenthal, R.L.; Wiedmann, M.; Dowd, S.E.; et al. Equine stomachs harbor an abundant and diverse mucosal microbiota. Appl. Environ. Microbiol. 2012, 78, 2522–2532. [Google Scholar] [CrossRef] [PubMed]
- Yuki, N.; Shimazaki, T.; Kushiro, A.; Watanabe, K.; Uchida, K.; Yuyama, T.; Morotomi, M. Colonization of the stratified squamous epithelium of the nonsecreting area of horse stomach by lactobacilli. Appl. Environ. Microbiol. 2000, 66, 5030–5034. [Google Scholar] [CrossRef] [PubMed]
- Dougal, K.; De La Fuente, G.; Harris, P.A.; Girdwood, S.E.; Pinloche, E.; Newbold, C.J. Identification of a core bacterial community within the large intestine of the horse. PLoS ONE 2013, 8, e77660. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Tsolis, R.M.; Bäumler, A.J. The microbiome and gut homeostasis. Science 2022, 377, eabp9960. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E.S.; Bittinger, K.; Esipova, T.V.; Hou, L.; Chau, L.; Jiang, J.; Mesaros, C.; Lund, P.J.; Liang, X.; FitzGerald, G.A.; et al. Microbes vs. chemistry in the origin of the anaerobic gut lumen. Proc. Natl. Acad. Sci. USA 2018, 115, 4170–4175. [Google Scholar] [CrossRef]
- Fan, P.; Liu, P.; Song, P.; Chen, X.; Ma, X. Moderate dietary protein restriction alters the composition of gut microbiota and improves ileal barrier function in adult pig model. Sci. Rep. 2017, 7, 43412. [Google Scholar] [CrossRef]
- Morrison, P.K.; Newbold, C.J.; Jones, E.; Worgan, H.J.; Grove-White, D.H.; Dugdale, A.H.; Barfoot, C.; Harris, P.A.; Argo, C.M. The Equine Gastrointestinal Microbiome: Impacts of Age and Obesity. Front. Microbiol 2018, 9, 3017. [Google Scholar] [CrossRef]
- Dougal, K.; La Fuente, G.d.; Harris, P.A.; Girdwood, S.E.; Pinloche, E.; Geor, R.J.; Nielsen, B.D.; Schott, H.C.; Elzinga, S.; Newbold, C.J. Characterisation of the faecal bacterial community in adult and elderly horses fed a high fibre, high oil or high starch diet using 454 pyrosequencing. PLoS ONE 2014, 9, e87424. [Google Scholar] [CrossRef]
- Costa, M.C.; Stämpfli, H.R.; Allen-Vercoe, E.; Weese, J.S. Development of the faecal microbiota in foals. Equine Vet. J. 2016, 48, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Lindenberg, F.; Krych, L.; Kot, W.; Fielden, J.; Frøkiær, H.; van Galen, G.; Nielsen, D.S.; Hansen, A.K. Development of the equine gut microbiota. Sci. Rep. 2019, 9, 14427. [Google Scholar] [CrossRef]
- Pryde, S.E.; Duncan, S.H.; Hold, G.L.; Stewart, C.S.; Flint, H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 2002, 217, 133–139. [Google Scholar] [CrossRef]
- Lindenberg, F.; Krych, L.; Fielden, J.; Kot, W.; Frøkiær, H.; van Galen, G.; Nielsen, D.S.; Hansen, A.K. Expression of immune regulatory genes correlate with the abundance of specific Clostridiales and Verrucomicrobia species in the equine ileum and cecum. Sci. Rep. 2019, 9, 12674. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.A.; Kittelmann, S.; Rogers, C.W.; Gee, E.K.; Bolwell, C.F.; Bermingham, E.N.; Thomas, D.G. Faecal microbiota of forage-fed horses in New Zealand and the population dynamics of microbial communities following dietary change. PLoS ONE 2014, 9, e112846. [Google Scholar] [CrossRef]
- O’ Donnell, M.M.; Harris, H.M.B.; Jeffery, I.B.; Claesson, M.J.; Younge, B.; O’ Toole, P.W.; Ross, R.P. The core faecal bacterial microbiome of Irish Thoroughbred racehorses. Lett. Appl. Microbiol. 2013, 57, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Proudman, C.J.; Hunter, J.O.; Darby, A.C.; Escalona, E.E.; Batty, C.; Turner, C. Characterisation of the faecal metabolome and microbiome of Thoroughbred racehorses. Equine Vet. J. 2015, 47, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Taminiau, B.; Brévers, B.; Avesani, V.; van Broeck, J.; Leroux, A.; Gallot, M.; Bruwier, A.; Amory, H.; Delmée, M.; et al. Faecal microbiota characterisation of horses using 16 rdna barcoded pyrosequencing, and carriage rate of clostridium difficile at hospital admission. BMC Microbiol. 2015, 15, 181. [Google Scholar] [CrossRef]
- Willing, B.; Vörös, A.; Roos, S.; Jones, C.; Jansson, A.; Lindberg, J.E. Changes in faecal bacteria associated with concentrate and forage-only diets fed to horses in training. Equine Vet. J. 2009, 41, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, B.; Bai, D.; Huang, J.; Shiraigo, W.; Yang, L.; Zhao, Q.; Ren, X.; Wu, J.; Bao, W.; et al. Comparison of Fecal Microbiota of Mongolian and Thoroughbred Horses by High-throughput Sequencing of the V4 Region of the 16S rRNA Gene. Asian-Australas. J. Anim. Sci. 2016, 29, 1345–1352. [Google Scholar] [CrossRef]
- Costa, M.C.; Arroyo, L.G.; Allen-Vercoe, E.; Stämpfli, H.R.; Kim, P.T.; Sturgeon, A.; Weese, J.S. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3–V5 region of the 16S rRNA gene. PLoS ONE 2012, 7, e41484. [Google Scholar] [CrossRef]
- Shepherd, M.L.; Swecker, W.S.; Jensen, R.V.; Ponder, M.A. Characterization of the fecal bacteria communities of forage-fed horses by pyrosequencing of 16S rRNA V4 gene amplicons. FEMS Microbiol. Lett. 2012, 326, 62–68. [Google Scholar] [CrossRef]
- Weese, J.S.; Holcombe, S.J.; Embertson, R.M.; Kurtz, K.A.; Roessner, H.A.; Jalali, M.; Wismer, S.E. Changes in the faecal microbiota of mares precede the development of post partum colic. Equine Vet. J. 2015, 47, 641–649. [Google Scholar] [CrossRef]
- Steelman, S.M.; Chowdhary, B.P.; Dowd, S.; Suchodolski, J.; Janečka, J.E. Pyrosequencing of 16S rRNA genes in fecal samples reveals high diversity of hindgut microflora in horses and potential links to chronic laminitis. BMC Vet. Res. 2012, 8, 231. [Google Scholar] [CrossRef]
- McKinney, C.A.; Oliveira, B.C.M.; Bedenice, D.; Paradis, M.-R.; Mazan, M.; Sage, S.; Sanchez, A.; Widmer, G. The fecal microbiota of healthy donor horses and geriatric recipients undergoing fecal microbial transplantation for the treatment of diarrhea. PLoS ONE 2020, 15, e0230148. [Google Scholar] [CrossRef]
- Yu, Z.; García-González, R.; Schanbacher, F.L.; Morrison, M. Evaluations of different hypervariable regions of archaeal 16S rRNA genes in profiling of methanogens by Archaea-specific PCR and denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 2008, 74, 889–893. [Google Scholar] [CrossRef]
- Franz, R.; Soliva, C.R.; Kreuzer, M.; Hummel, J.; Clauss, M. Methane output of rabbits (Oryctolagus cuniculus) and guinea pigs (Cavia porcellus) fed a hay-only diet: Implications for the scaling of methane production with body mass in non-ruminant mammalian herbivores. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2011, 158, 177–181. [Google Scholar] [CrossRef]
- Daly, K.; Proudman, C.J.; Duncan, S.H.; Flint, H.J.; Dyer, J.; Shirazi-Beechey, S.P. Alterations in microbiota and fermentation products in equine large intestine in response to dietary variation and intestinal disease. Br. J. Nutr. 2012, 107, 989–995. [Google Scholar] [CrossRef]
- Rampelli, S.; Guenther, K.; Turroni, S.; Wolters, M.; Veidebaum, T.; Kourides, Y.; Molnár, D.; Lissner, L.; Benitez-Paez, A.; Sanz, Y.; et al. Pre-obese children’s dysbiotic gut microbiome and unhealthy diets may predict the development of obesity. Commun. Biol. 2018, 1, 222. [Google Scholar] [CrossRef]
- Li, L.; Su, Q.; Xie, B.; Duan, L.; Zhao, W.; Hu, D.; Wu, R.; Liu, H. Gut microbes in correlation with mood: Case study in a closed experimental human life support system. Neurogastroenterol. Motil. 2016, 28, 1233–1240. [Google Scholar] [CrossRef]
- Panasevich, M.R.; Morris, E.M.; Chintapalli, S.V.; Wankhade, U.D.; Shankar, K.; Britton, S.L.; Koch, L.G.; Thyfault, J.P.; Rector, R.S. Gut microbiota are linked to increased susceptibility to hepatic steatosis in low-aerobic-capacity rats fed an acute high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G166–G179. [Google Scholar] [CrossRef]
- Tuniyazi, M.; He, J.; Guo, J.; Li, S.; Zhang, N.; Hu, X.; Fu, Y. Changes of microbial and metabolome of the equine hindgut during oligofructose-induced laminitis. BMC Vet. Res. 2021, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.E.; Shetty, S.A.; van den Berg, P.; Burden, F.; van Doorn, D.A.; Pellikaan, W.F.; Dijkstra, J.; Smidt, H. Multi-kingdom characterization of the core equine fecal microbiota based on multiple equine (sub)species. Anim. Microbiome 2020, 2, 6. [Google Scholar] [CrossRef]
- Mackie, R.I.; Wilkins, C.A. Enumeration of anaerobic bacterial microflora of the equine gastrointestinal tract. Appl. Environ. Microbiol. 1988, 54, 2155–2160. [Google Scholar] [CrossRef] [PubMed]
- Massacci, F.R.; Clark, A.; Ruet, A.; Lansade, L.; Costa, M.; Mach, N. Inter-breed diversity and temporal dynamics of the faecal microbiota in healthy horses. J. Anim. Breed. Genet. 2019, 137, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Quercia, S.; Freccero, F.; Castagnetti, C.; Soverini, M.; Turroni, S.; Biagi, E.; Rampelli, S.; Lanci, A.; Mariella, J.; Chinellato, E.; et al. Early colonisation and temporal dynamics of the gut microbial ecosystem in Standardbred foals. Equine Vet. J. 2019, 51, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Salem, S.E.; Maddox, T.W.; Berg, A.; Antczak, P.; Ketley, J.M.; Williams, N.J.; Archer, D.C. Variation in faecal microbiota in a group of horses managed at pasture over a 12-month period. Sci. Rep. 2018, 8, 8510. [Google Scholar] [CrossRef]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129. [Google Scholar] [CrossRef]
- Jiménez, E.; Marín, M.L.; Martín, R.; Odriozola, J.M.; Olivares, M.; Xaus, J.; Fernández, L.; Rodríguez, J.M. Is meconium from healthy newborns actually sterile? Res. Microbiol. 2008, 159, 187–193. [Google Scholar] [CrossRef]
- Perez-Muñoz, M.E.; Arrieta, M.-C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef]
- La Torre, U.d.; Henderson, J.D.; Furtado, K.L.; Pedroja, M.; Elenamarie, O.M.; Mora, A.; Pechanec, M.Y.; Maga, E.A.; Mienaltowski, M.J. Utilizing the fecal microbiota to understand foal gut transitions from birth to weaning. PLoS ONE 2019, 14, e0216211. [Google Scholar] [CrossRef]
- Husso, A.; Jalanka, J.; Alipour, M.J.; Huhti, P.; Kareskoski, M.; Pessa-Morikawa, T.; Iivanainen, A.; Niku, M. The composition of the perinatal intestinal microbiota in horse. Sci. Rep. 2020, 10, 441. [Google Scholar] [CrossRef]
- Yang, Q.; Huang, X.; Zhao, S.; Sun, W.; Yan, Z.; Wang, P.; Li, S.; Huang, W.; Zhang, S.; Liu, L.; et al. Structure and Function of the Fecal Microbiota in Diarrheic Neonatal Piglets. Front. Microbiol. 2017, 8, 502. [Google Scholar] [CrossRef] [PubMed]
- Gresse, R.; Chaucheyras Durand, F.; Dunière, L.; Blanquet-Diot, S.; Forano, E. Microbiota Composition and Functional Profiling Throughout the Gastrointestinal Tract of Commercial Weaning Piglets. Microorganisms 2019, 7, 343. [Google Scholar] [CrossRef] [PubMed]
- Yeoman, C.J.; Ishaq, S.L.; Bichi, E.; Olivo, S.K.; Lowe, J.; Aldridge, B.M. Biogeographical Differences in the Influence of Maternal Microbial Sources on the Early Successional Development of the Bovine Neonatal Gastrointestinal tract. Sci. Rep. 2018, 8, 3197. [Google Scholar] [CrossRef] [PubMed]
- Freestone, P.; Lyte, M. Stress and microbial endocrinology: Prospects for ruminant nutrition. Animal 2010, 4, 1248–1257. [Google Scholar] [CrossRef]
- Lyte, M. Probiotics function mechanistically as delivery vehicles for neuroactive compounds: Microbial endocrinology in the design and use of probiotics. BioEssays News Rev. Mol. Cell. Dev. Biol. 2011, 33, 574–581. [Google Scholar] [CrossRef]
- Lyte, M. Microbial endocrinology in the microbiome-gut-brain axis: How bacterial production and utilization of neurochemicals influence behavior. PLoS Pathog. 2013, 9, e1003726. [Google Scholar] [CrossRef]
- Mach, N.; Foury, A.; Kittelmann, S.; Reigner, F.; Moroldo, M.; Ballester, M.; Esquerré, D.; Rivière, J.; Sallé, G.; Gérard, P.; et al. The Effects of Weaning Methods on Gut Microbiota Composition and Horse Physiology. Front. Physiol 2017, 8, 535. [Google Scholar] [CrossRef]
- Mura, E.; Edwards, J.; Kittelmann, S.; Kaerger, K.; Voigt, K.; Mrázek, J.; Moniello, G.; Fliegerova, K. Anaerobic fungal communities differ along the horse digestive tract. Fungal Biol. 2019, 123, 240–246. [Google Scholar] [CrossRef]
- Medina, B.; Girard, I.D.; Jacotot, E.; Julliand, V. Effect of a preparation of Saccharomyces cerevisiae on microbial profiles and fermentation patterns in the large intestine of horses fed a high fiber or a high starch diet. J. Anim. Sci. 2002, 80, 2600–2609. [Google Scholar] [CrossRef]
- Jouany, J.-P.; Medina, B.; Bertin, G.; Julliand, V. Effect of live yeast culture supplementation on hindgut microbial communities and their polysaccharidase and glycoside hydrolase activities in horses fed a high-fiber or high-starch diet. J. Anim. Sci. 2009, 87, 2844–2852. [Google Scholar] [CrossRef] [PubMed]
- Plaizier, J.C.; Danesh Mesgaran, M.; Derakhshani, H.; Golder, H.; Khafipour, E.; Kleen, J.L.; Lean, I.; Loor, J.; Penner, G.; Zebeli, Q. Review: Enhancing gastrointestinal health in dairy cows. Anim. Int. J. Anim. Biosci. 2018, 12, s399–s418. [Google Scholar] [CrossRef] [PubMed]
- Jouany, J.-P.; Gobert, J.; Medina, B.; Bertin, G.; Julliand, V. Effect of live yeast culture supplementation on apparent digestibility and rate of passage in horses fed a high-fiber or high-starch diet. J. Anim. Sci. 2008, 86, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.; Hastie, P.; Murray, J.-A. Factors Influencing Equine Gut Microbiota: Current Knowledge. J. Equine Vet. Sci. 2020, 88, 102943. [Google Scholar] [CrossRef]
- Bulmer, L.S.; Murray, J.-A.; Burns, N.M.; Garber, A.; Wemelsfelder, F.; McEwan, N.R.; Hastie, P.M. High-starch diets alter equine faecal microbiota and increase behavioural reactivity. Sci. Rep. 2019, 9, 18621. [Google Scholar] [CrossRef]
- Grimm, P.; Combes, S.; Pascal, G.; Cauquil, L.; Julliand, V. Dietary composition and yeast/microalgae combination supplementation modulate the microbial ecosystem in the caecum, colon and faeces of horses. Br. J. Nutr. 2019, 1–27. [Google Scholar] [CrossRef]
- Morrison, P.K.; Newbold, C.J.; Jones, E.; Worgan, H.J.; Grove-White, D.H.; Dugdale, A.H.; Barfoot, C.; Harris, P.A.; Argo, C.M. The equine gastrointestinal microbiome: Impacts of weight-loss. BMC Vet. Res. 2020, 16, 78. [Google Scholar] [CrossRef] [PubMed]
- Destrez, A.; Grimm, P.; Julliand, V. Dietary-induced modulation of the hindgut microbiota is related to behavioral responses during stressful events in horses. Physiol. Behav. 2019, 202, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Destrez, A.; Grimm, P.; Cézilly, F.; Julliand, V. Changes of the hindgut microbiota due to high-starch diet can be associated with behavioral stress response in horses. Physiol. Behav. 2015, 149, 159–164. [Google Scholar] [CrossRef]
- Mach, N.; Ruet, A.; Clark, A.; Bars-Cortina, D.; Ramayo-Caldas, Y.; Crisci, E.; Pennarun, S.; Dhorne-Pollet, S.; Foury, A.; Moisan, M.-P.; et al. Priming for welfare: Gut microbiota is associated with equitation conditions and behavior in horse athletes. Sci. Rep. 2020, 10, 8311. [Google Scholar] [CrossRef]
- Venable, E.B.; Fenton, K.A.; Braner, V.M.; Reddington, C.E.; Halpin, M.J.; Heitz, S.A.; Francis, J.M.; Gulson, N.A.; Goyer, C.L.; Bland, S.D.; et al. Effects of Feeding Management on the Equine Cecal Microbiota. J. Equine Vet. Sci. 2017, 49, 113–121. [Google Scholar] [CrossRef]
- Perry, E.; Cross, T.-W.L.; Francis, J.M.; Holscher, H.D.; Clark, S.D.; Swanson, K.S. Effect of Road Transport on the Equine Cecal Microbiota. J. Equine Vet. Sci. 2018, 68, 12–20. [Google Scholar] [CrossRef]
- Padalino, B.; Raidal, S.L.; Hall, E.; Knight, P.; Celi, P.; Jeffcott, L.; Muscatello, G. Survey of horse transportation in Australia: Issues and practices. Aust. Vet. J. 2016, 94, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Faubladier, C.; Chaucheyras-Durand, F.; da Veiga, L.; Julliand, V. Effect of transportation on fecal bacterial communities and fermentative activities in horses: Impact of Saccharomyces cerevisiae CNCM I-1077 supplementation. J. Anim. Sci. 2013, 91, 1736–1744. [Google Scholar] [CrossRef]
- Allen, J.M.; Berg Miller, M.E.; Pence, B.D.; Whitlock, K.; Nehra, V.; Gaskins, H.R.; White, B.A.; Fryer, J.D.; Woods, J.A. Voluntary and forced exercise differentially alters the gut microbiome in C57BL/6J mice. J. Appl. Physiol. 2015, 118, 1059–1066. [Google Scholar] [CrossRef]
- Clark, A.; Mach, N. Exercise-induced stress behavior, gut-microbiota-brain axis and diet: A systematic review for athletes. J. Int. Soc. Sports Nutr. 2016, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Mach, N.; Moroldo, M.; Rau, A.; Lecardonnel, J.; Le Moyec, L.; Robert, C.; Barrey, E. Understanding the Holobiont: Crosstalk Between Gut Microbiota and Mitochondria During Long Exercise in Horse. Front. Mol. Biosci. 2021, 8, 656204. [Google Scholar] [CrossRef]
- Cymbaluk, N.F.; Christison, G.I. Environmental Effects on Thermoregulation and Nutrition of Horses. Vet. Clin. N. Am. Equine Pract. 1990, 6, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Tajima, K.; Nonaka, I.; Higuchi, K.; Takusari, N.; Kurihara, M.; Takenaka, A.; Mitsumori, M.; Kajikawa, H.; Aminov, R.I. Influence of high temperature and humidity on rumen bacterial diversity in Holstein heifers. Anaerobe 2007, 13, 57–64. [Google Scholar] [CrossRef]
- Zhong, S.; Ding, Y.; Wang, Y.; Zhou, G.; Guo, H.; Chen, Y.; Yang, Y. Temperature and humidity index (THI)-induced rumen bacterial community changes in goats. Appl. Microbiol. Biotechnol. 2019, 103, 3193–3203. [Google Scholar] [CrossRef]
- Zhao, S.; Min, L.; Zheng, N.; Wang, J. Effect of Heat Stress on Bacterial Composition and Metabolism in the Rumen of Lactating Dairy Cows. Animals 2019, 9, 925. [Google Scholar] [CrossRef] [PubMed]
- Uyeno, Y.; Sekiguchi, Y.; Tajima, K.; Takenaka, A.; Kurihara, M.; Kamagata, Y. An rRNA-based analysis for evaluating the effect of heat stress on the rumen microbial composition of Holstein heifers. Anaerobe 2010, 16, 27–33. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Guo, H.; Zheng, W.; Xue, Y.; Zhao, R.; Yao, W. Heat stress affects fecal microbial and metabolic alterations of primiparous sows during late gestation. J. Anim. Sci. Biotechnol. 2019, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Labussière, E.; Achard, C.; Dubois, S.; Combes, S.; Castex, M.; Renaudeau, D. Saccharomyces cerevisiae boulardii CNCM I-1079 supplementation in finishing male pigs helps to cope with heat stress through feeding behaviour and gut microbiota modulation. Br. J. Nutr. 2022, 127, 353–368. [Google Scholar] [CrossRef]
- Dong, H.-J.; Ho, H.; Hwang, H.; Kim, Y.; Han, J.; Lee, I.; Cho, S. Diversity of the Gastric Microbiota in Thoroughbred Racehorses Having Gastric Ulcer. J. Microbiol. Biotechnol. 2016, 26, 763–774. [Google Scholar] [CrossRef]
- Paul, L.J.; Ericsson, A.C.; Andrews, F.M.; Keowen, M.L.; Morales Yniguez, F.; Garza, F.; Banse, H.E. Gastric microbiome in horses with and without equine glandular gastric disease. J. Vet. Intern. Med. 2021, 35, 2458–2464. [Google Scholar] [CrossRef]
- Voss, S.J.; McGuinness, D.H.; Weir, W.; Sutton, D.G.M. A study comparing the healthy and diseased equine glandular gastric microbiota sampled with sheathed transendoscopic cytology brushes. J. Equine Vet. Sci. 2022, 116, 104002. [Google Scholar] [CrossRef]
- Arroyo, L.G.; Rossi, L.; Santos, B.P.; Gomez, D.E.; Surette, M.G.; Costa, M.C. Luminal and Mucosal Microbiota of the Cecum and Large Colon of Healthy and Diarrheic Horses. Animals 2020, 10, 1403. [Google Scholar] [CrossRef]
- Rivera-Chávez, F.; Zhang, L.F.; Faber, F.; Lopez, C.A.; Byndloss, M.X.; Olsan, E.E.; Xu, G.; Velazquez, E.M.; Lebrilla, C.B.; Winter, S.E.; et al. Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe 2016, 19, 443–454. [Google Scholar] [CrossRef]
- Park, T.; Cheong, H.; Yoon, J.; Kim, A.; Yun, Y.; Unno, T. Comparison of the Fecal Microbiota of Horses with Intestinal Disease and Their Healthy Counterparts. Vet. Sci. 2021, 8, 113. [Google Scholar] [CrossRef]
- Stewart, H.L.; Pitta, D.; Indugu, N.; Vecchiarelli, B.; Hennessy, M.L.; Engiles, J.B.; Southwood, L.L. Changes in the faecal bacterial microbiota during hospitalisation of horses with colic and the effect of different causes of colic. Equine Vet. J. 2021, 53, 1119–1131. [Google Scholar] [CrossRef]
- Salem, S.E.; Hough, R.; Probert, C.; Maddox, T.W.; Antczak, P.; Ketley, J.M.; Williams, N.J.; Stoneham, S.J.; Archer, D.C. A longitudinal study of the faecal microbiome and metabolome of periparturient mares. PeerJ 2019, 7, e6687. [Google Scholar] [CrossRef]
- Lindroth, K.M.; Johansen, A.; Båverud, V.; Dicksved, J.; Lindberg, J.E.; Müller, C.E. Differential Defecation of Solid and Liquid Phases in Horses—A Descriptive Survey. Animals 2020, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, L.; Edwards, J.E.; Hermes, G.D.A.; Lúthersson, N.; van Doorn, D.A.; Okrathok, S.; Kujawa, T.J.; Smidt, H. Free Faecal Water: Analysis of Horse Faecal Microbiota and the Impact of Faecal Microbial Transplantation on Symptom Severity. Animals 2021, 11, 2776. [Google Scholar] [CrossRef] [PubMed]
- Schoster, A.; Weese, J.S.; Gerber, V.; Nicole Graubner, C. Dysbiosis is not present in horses with fecal water syndrome when compared to controls in spring and autumn. J. Vet. Intern. Med. 2020, 34, 1614–1621. [Google Scholar] [CrossRef]
- Kienzle, E.; Zehnder, C.; Pfister, K.; Gerhards, H.; Sauter-Louis, C.; Harris, P. Field Study on Risk Factors for Free Fecal Water in Pleasure Horses. J. Equine Vet. Sci. 2016, 44, 32–36. [Google Scholar] [CrossRef]
- Clark, A.; Sallé, G.; Ballan, V.; Reigner, F.; Meynadier, A.; Cortet, J.; Koch, C.; Riou, M.; Blanchard, A.; Mach, N. Strongyle Infection and Gut Microbiota: Profiling of Resistant and Susceptible Horses Over a Grazing Season. Front. Physiol. 2018, 9, 272. [Google Scholar] [CrossRef]
- Midha, A.; Schlosser, J.; Hartmann, S. Reciprocal Interactions between Nematodes and Their Microbial Environments. Front. Cell. Infect. Microbiol. 2017, 7, 144. [Google Scholar] [CrossRef]
- Peachey, L.E.; Castro, C.; Molena, R.A.; Jenkins, T.P.; Griffin, J.L.; Cantacessi, C. Dysbiosis associated with acute helminth infections in herbivorous youngstock—Observations and implications. Sci. Rep. 2019, 9, 11121. [Google Scholar] [CrossRef]
- Peachey, L.E.; Molena, R.A.; Jenkins, T.P.; Di Cesare, A.; Traversa, D.; Hodgkinson, J.E.; Cantacessi, C. The relationships between faecal egg counts and gut microbial composition in UK Thoroughbreds infected by cyathostomins. Int. J. Parasitol. 2018, 48, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Sallé, G.; Cortet, J.; Bois, I.; Dubès, C.; Guyot-Sionest, Q.; Larrieu, C.; Landrin, V.; Majorel, G.; Wittreck, S.; Woringer, E.; et al. Risk factor analysis of equine strongyle resistance to anthelmintics. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Goachet, A.G.; Ricard, J.M.; Jacotot, E.; Varloud, M.; Julliand, V. Effet de l’administration orale de trois anthelminthiques sur microflore colique cheval. Les Journées AVEF 2004, 548–550. [Google Scholar]
- Kunz, I.G.Z.; Reed, K.J.; Metcalf, J.L.; Hassel, D.M.; Coleman, R.J.; Hess, T.M.; Coleman, S.J. Equine Fecal Microbiota Changes Associated with Anthelmintic Administration. J. Equine Vet. Sci. 2019, 77, 98–106. [Google Scholar] [CrossRef]
- Rodiles, A.; Sacy, A.; Apper, E. Investigation of Anthelmintic Administration on the Equine Faecal Microbiota Abstract and poster. In Proceedings of the 9th Beneficial Microbes Conference, Amsterdam, The Netherlands, November 14–16 2022. [Google Scholar]
- Frank, N.; Geor, R.J.; Bailey, S.R.; Durham, A.E.; Johnson, P.J. Equine metabolic syndrome. J. Vet. Intern. Med. 2010, 24, 467–475. [Google Scholar] [CrossRef]
- Durham, A.E.; Frank, N.; McGowan, C.M.; Menzies-Gow, N.J.; Roelfsema, E.; Vervuert, I.; Feige, K.; Fey, K. ECEIM consensus statement on equine metabolic syndrome. J. Vet. Intern. Med. 2019, 33, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Biddle, A.S.; Tomb, J.-F.; Fan, Z. Microbiome and Blood Analyte Differences Point to Community and Metabolic Signatures in Lean and Obese Horses. Front. Vet. Sci. 2018, 5, 225. [Google Scholar] [CrossRef]
- Wyse, C.A.; McNie, K.A.; Tannahill, V.J.; Murray, J.K.; Love, S. Prevalence of obesity in riding horses in Scotland. Vet. Rec. 2008, 162, 590–591. [Google Scholar] [CrossRef]
- Stephenson, H.M.; Green, M.J.; Freeman, S.L. Prevalence of obesity in a population of horses in the UK. Vet. Rec. 2011, 168, 131. [Google Scholar] [CrossRef]
- Thatcher, C.D.; Pleasant, R.S.; Geor, R.J.; Elvinger, F. Prevalence of overconditioning in mature horses in southwest Virginia during the summer. J. Vet. Intern. Med. 2012, 26, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.; Keen, J.; McGowan, C. Equine metabolic syndrome. Vet. Rec. 2015, 177, 173–179. [Google Scholar] [CrossRef]
- Millinovich, G.J.; Trott, D.J.; Burrell, P.C.; Croser, E.L.; Al Jassim, R.A.M.; Morton, J.M.; van Eps, A.W.; Pollitt, C.C. Fluorescence in situ hybridization analysis of hindgut bacteria associated with the development of equine laminitis. Environ. Microbiol. 2007, 9, 2090–2100. [Google Scholar] [CrossRef]
- Elzinga, S.E.; Betancourt, A.; Stewart, J.C.; Altman, M.H.; Barker, V.D.; Muholland, M.; Bailey, S.; Brennan, K.M.; Adams, A.A. Effects of Docosahexaenoic Acid-Rich Microalgae Supplementation on Metabolic and Inflammatory Parameters in Horses with Equine Metabolic Syndrome. J. Equine Vet. Sci. 2019, 83, 102811. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.R.; Marr, C.M.; Elliott, J. Identification and quantification of amines in the equine caecum. Res. Vet. Sci. 2003, 74, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Al Jassim, R.A.M.; Scott, P.T.; Trebbin, A.L.; Trott, D.; Pollitt, C.C. The genetic diversity of lactic acid producing bacteria in the equine gastrointestinal tract. FEMS Microbiol. Lett. 2005, 248, 75–81. [Google Scholar] [CrossRef]
- Kaneko, T.; Bando, Y.; Kurihara, H.; Satomi, K.; Nonoyama, K.; Matsuura, N. Fecal microflora in a patient with short-bowel syndrome and identification of dominant lactobacilli. J. Clin. Microbiol. 1997, 35, 3181–3185. [Google Scholar] [CrossRef] [PubMed]
- Garner, M.R.; Flint, J.F.; Russell, J.B. Allisonella histaminiformans gen. nov., sp. nov. A novel bacterium that produces histamine, utilizes histidine as its sole energy source, and could play a role in bovine and equine laminitis. Syst. Appl. Microbiol. 2002, 25, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.M.; Eades, S.C.; Reinemeyer, C.R.; Fugaro, M.N.; Onishi, J.C. Illumina sequencing of the V4 hypervariable region 16S rRNA gene reveals extensive changes in bacterial communities in the cecum following carbohydrate oral infusion and development of early-stage acute laminitis in the horse. Vet. Microbiol. 2014, 168, 436–441. [Google Scholar] [CrossRef]
- Fitzgerald, D.M.; Spence, R.J.; Stewart, Z.K.; Prentis, P.J.; Sillence, M.N.; de Laat, M.A. The effect of diet change and insulin dysregulation on the faecal microbiome of ponies. J. Exp. Biol. 2020, 223, jeb219154. [Google Scholar] [CrossRef]
- Langner, K.; Blaue, D.; Schedlbauer, C.; Starzonek, J.; Julliand, V.; Vervuert, I. Changes in the faecal microbiota of horses and ponies during a two-year body weight gain programme. PLoS ONE 2020, 15, e0230015. [Google Scholar] [CrossRef]
- Dougal, K.; Harris, P.A.; Girdwood, S.E.; Creevey, C.J.; Curtis, G.C.; Barfoot, C.F.; Argo, C.M.; Newbold, C.J. Changes in the Total Fecal Bacterial Population in Individual Horses Maintained on a Restricted Diet over 6 Weeks. Front. Microbiol. 2017, 8, 1502. [Google Scholar] [CrossRef]
- Coleman, M.C.; Whitfield-Cargile, C.M.; Madrigal, R.G.; Cohen, N.D. Comparison of the microbiome, metabolome, and lipidome of obese and non-obese horses. PLoS ONE 2019, 14, e0215918. [Google Scholar] [CrossRef] [PubMed]
- Walshe, N.; Cabrera-Rubio, R.; Collins, R.; Puggioni, A.; Gath, V.; Crispie, F.; Cotter, P.D.; Brennan, L.; Mulcahy, G.; Duggan, V. A Multiomic Approach to Investigate the Effects of a Weight Loss Program on the Intestinal Health of Overweight Horses. Front. Vet. Sci. 2021, 8, 668120. [Google Scholar] [CrossRef]
- Nagano, T.; Yano, H. Dietary cellulose nanofiber modulates obesity and gut microbiota in high-fat-fed mice. Bioact. Carbohydr. Diet. Fibre 2020, 22, 100214. [Google Scholar] [CrossRef]
- Zak, A.; Siwinska, N.; Elzinga, S.; Barker, V.D.; Stefaniak, T.; Schanbacher, B.J.; Place, N.J.; Niedzwiedz, A.; Adams, A.A. Effects of equine metabolic syndrome on inflammation and acute-phase markers in horses. Domest. Anim. Endocrinol. 2020, 72, 106448. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Tsolis, R.M.; Bäumler, A.J. Dysbiotic Proteobacteria expansion: A microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Abot, A.; Fried, S.; Cani, P.D.; Knauf, C. Reactive Oxygen Species/Reactive Nitrogen Species as Messengers in the Gut: Impact on Physiology and Metabolic Disorders. Antioxid. Redox Signal. 2022, 37, 394–415. [Google Scholar] [CrossRef]
- Rath, E.; Haller, D. Intestinal epithelial cell metabolism at the interface of microbial dysbiosis and tissue injury. Mucosal Immunol. 2022, 15, 595–604. [Google Scholar] [CrossRef]
- Steinberg, L.M.; Regan, J.M. mcrA-targeted real-time quantitative PCR method to examine methanogen communities. Appl. Environ. Microbiol. 2009, 75, 4435–4442. [Google Scholar] [CrossRef] [PubMed]
- Comtet-Marre, S.; Chaucheyras-Durand, F.; Bouzid, O.; Mosoni, P.; Bayat, A.R.; Peyret, P.; Forano, E. FibroChip, a Functional DNA Microarray to Monitor Cellulolytic and Hemicellulolytic Activities of Rumen Microbiota. Front. Microbiol. 2018, 9, 215. [Google Scholar] [CrossRef]
- Turroni, F.; Ventura, M.; Buttó, L.F.; Duranti, S.; O’Toole, P.W.; Motherway, M.O.C.; van Sinderen, D. Molecular dialogue between the human gut microbiota and the host: A Lactobacillus and Bifidobacterium perspective. Cell. Mol. Life Sci. 2014, 71, 183–203. [Google Scholar] [CrossRef]
- Harlow, B.E.; Lawrence, L.M.; Harris, P.A.; Aiken, G.E.; Flythe, M.D. Exogenous lactobacilli mitigate microbial changes associated with grain fermentation (corn, oats, and wheat) by equine fecal microflora ex vivo. PLoS ONE 2017, 12, e0174059. [Google Scholar] [CrossRef] [PubMed]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Chaucheyras-Durand, F.; Dunière, L. The use of probiotics as supplements for ruminants. In Improving Rumen Function; McSweeney, C.S., Mackie, R.I., Eds.; Burleigh Dodds Science Publishing: Cambridge, UK, 2020; pp. 775–818. ISBN 9781786763327. [Google Scholar]
- Coverdale, J.A. HORSE SPECIES SYMPOSIUM: Can the microbiome of the horse be altered to improve digestion? J. Anim. Sci. 2016, 94, 2275–2281. [Google Scholar] [CrossRef] [PubMed]
- Palade, C.-M.; Vulpoi, G.-A.; Vulpoi, R.-A.; Drug, V.L.; Barboi, O.-B.; Ciocoiu, M. The Biotics Family: Current Knowledge and Future Perspectives in Metabolic Diseases. Life 2022, 12, 1263. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Cordoba, B.; Castro-López, C.; García, H.S.; González-Córdova, A.F.; Hernández-Mendoza, A. Postbiotics and paraprobiotics: A review of current evidence and emerging trends. Adv. Food Nutr. Res. 2020, 94, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.; Hastie, P.M.; Farci, V.; Bulmer, L.; Alzahal, O.; Murray, J.M.D. The effect of supplementing pony diets with yeast on 1. In vivo and in vitro digestibility, faecal pH and particle size. Anim. Int. J. Anim. Biosci. 2020, 14, 2481–2492. [Google Scholar] [CrossRef] [PubMed]
- Russouw, A.; Chevaux, E.; Chaucheyras-Durand, F.; Esposito, G.; Raffrenato, E. Effects of Saccharomyces cerevisiae, medium and forage type and their interactions on in vitro ruminal fermentation. Heliyon 2020, 6, e05028. [Google Scholar] [CrossRef]
- Tassone, S.; Renna, M.; Barbera, S.; Valle, E.; Fortina, R. In Vitro Digestibility Measurement of Feedstuffs in Donkeys Using the DaisyII Incubator. J. Equine Vet. Sci. 2019, 75, 122–126. [Google Scholar] [CrossRef]
- Lattimer, J.M.; Cooper, S.R.; Freeman, D.W.; Lalman, D.L. Effect of yeast culture on in vitro fermentation of a high-concentrate or high-fiber diet using equine fecal inoculum in a Daisy II incubator. J. Anim. Sci. 2007, 85, 2484–2491. [Google Scholar] [CrossRef]
- Tassone, S.; Fortina, R.; Peiretti, P.G. In Vitro Techniques Using the DaisyII Incubator for the Assessment of Digestibility: A Review. Animals 2020, 10, 775. [Google Scholar] [CrossRef]
- Leng, J.; Walton, G.; Swann, J.; Darby, A.; La Ragione, R.; Proudman, C. “Bowel on the Bench”: Proof of Concept of a Three-Stage, In Vitro Fermentation Model of the Equine Large Intestine. Appl. Environ. Microbiol. 2019, 86, e02093-19. [Google Scholar] [CrossRef] [PubMed]
- Gresse, R.; Chaucheyras-Durand, F.; Denis, S.; Beaumont, M.; van de Wiele, T.; Forano, E.; Blanquet-Diot, S. Weaning-associated feed deprivation stress causes microbiota disruptions in a novel mucin-containing in vitro model of the piglet colon (MPigut-IVM). J. Anim. Sci. Biotechnol. 2021, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Cordonnier, C.; Thévenot, J.; Etienne-Mesmin, L.; Denis, S.; Alric, M.; Livrelli, V.; Blanquet-Diot, S. Dynamic In Vitro Models of the Human Gastrointestinal Tract as Relevant Tools to Assess the Survival of Probiotic Strains and Their Interactions with Gut Microbiota. Microorganisms 2015, 3, 725–745. [Google Scholar] [CrossRef] [PubMed]







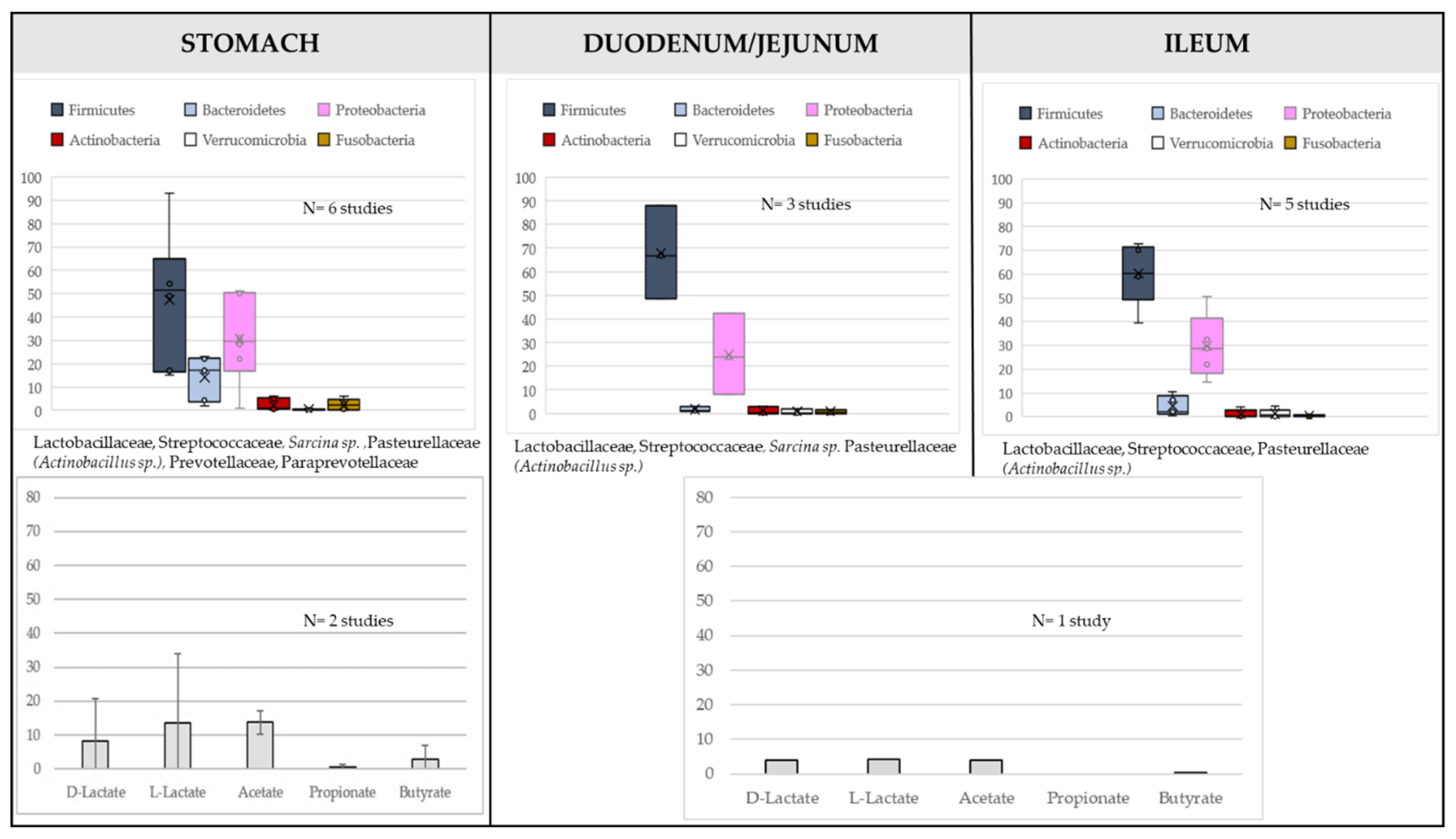{kind=link}
{kind=link}
{kind=link}
{kind=link}
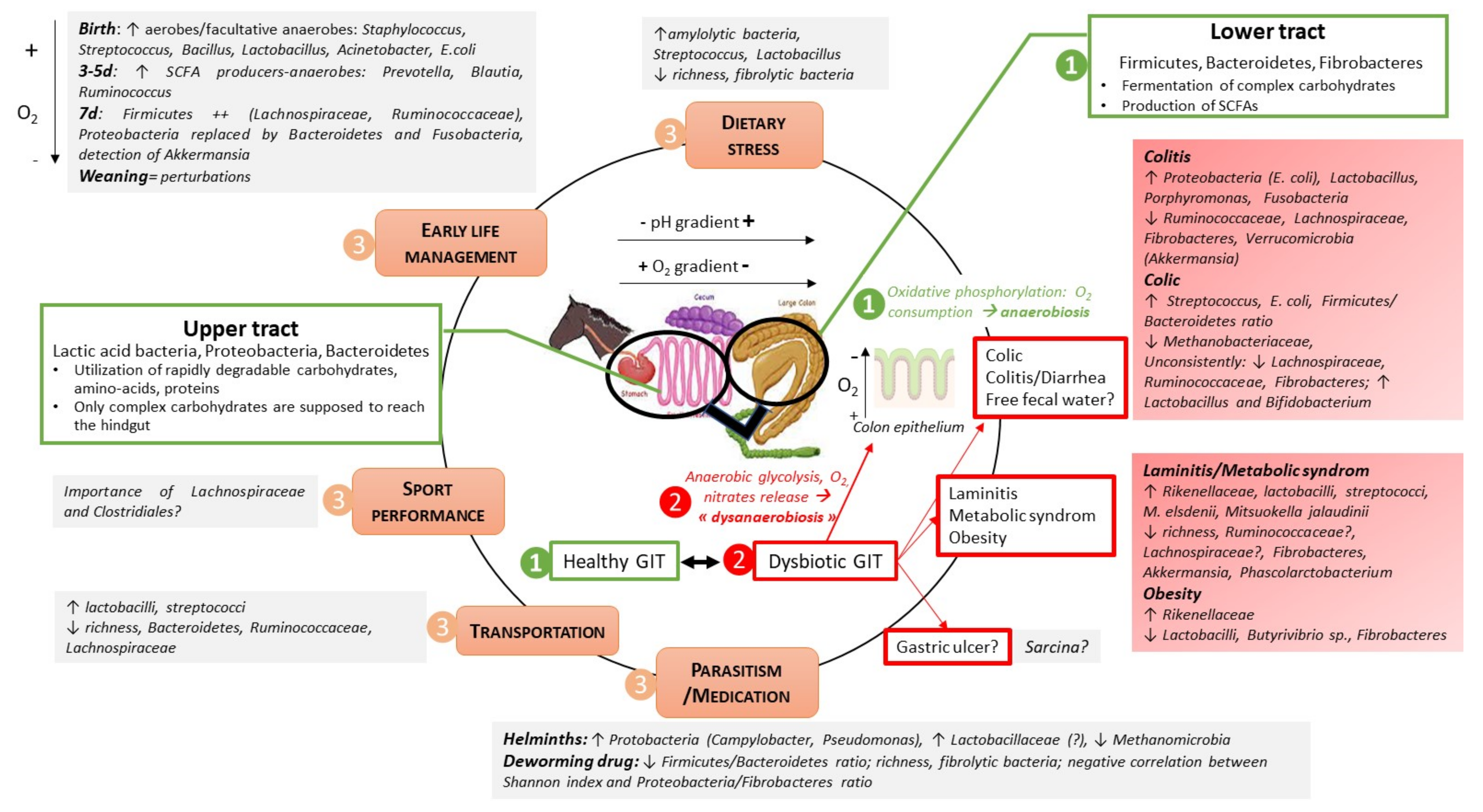{kind=link}
{kind=link}
| Reference | Type of Diet and Protocol Details | Effect on Microbiota |
|---|---|---|
| [61] | 4 fistulated horses in a 4 × 4 latin square design. Diets fed 2×/day. HF = 11.6% starch; 41% NDF HS = 30.1% starch; 30.7% NDF NDF:starch ratio of 3.5 for HF and 1.0 for HS diets Enumeration of cultivable viable functional bacterial groups in the cecum and colon | Increase in Lactobacilli in the cecum and the colon with HS diet but no change in total anaerobes, cellulolytics or streptococci |
| [60] | 8 fistulated horses in a 4 × 4 latin square design. Diets fed 2×/day HS = 3.4 g/kg BW of starch per meal but maintaining NDF:starch ratio of 1.0 Enumeration of cultivable viable functional bacterial groups in the cecum and colon | With HS, total anaerobic and lactic acid-utilizing bacteria increased, and cellulolytic bacteria decreased in the cecum. Increase in lactobacilli and streptococci both in the cecal and colonic contents |
| [19] | 17 mares Hay diet vs. hay plus a high cereal supplement (35% of starch in the high starch diet) 16S rDNA sequencing | With starch diet, increase in Proteobacteria phylum (Succinivibrio/Succinivibrionaceae related OTUs) Increase in Phocaeicola related OTU (Bacteroidetes phylum), increase in some Lachnospiraceae related OTUs but decrease in other Lachnospiraceae related OTUs (Firmicutes phylum) |
| [66] | 6 fistulated geldings in a 2 × 2 latin square design. Diets fed HS = 56%/44% hay/barley diet for 3 weeks (0.20% BW of starch per meal) HF = 100% hay 16S rDNA sequencing Enumeration of cultivable viable functional bacterial groups in the cecum and colon | Reduced bacterial diversity with HS Impact of HS diet on community composition: decrease in Ruminoclostridium genus in the cecum, decrease in Bacteroidales S24-7 and, Lachnospiraceae NC2004 groups, increase in Veillonellaceae family in the colon Total anaerobes, starch utilizers, lactate utilisers increased, and cellulose utilizers decreased |
| [65] | Ten 18-month-old ponies in a 2 × 2 cross-over design with 2 experimental diets HF and HS. Diets fed 2×/day. HF = hay and lucerne; 0.46 g/kg of BW of starch per meal HS = hay and compound mix; 0.96 g/kg BW of starch per meal 16S rDNA sequencing in the feces | Bacterial diversity lower in HS diet with higher variance Impact of HS diet on community composition: decrease in Ruminococcaceae family abundance and increase in Streptococcus OTU |
| [18] | 23 pony mares of different ages followed for 2 years HF: Hay diet at 2% body mass as daily dry matter intake for 4 weeks HS: 2 g starch per kg body mass distributed for maximum 5 days 16S rDNA sequencing in the feces | Diet transition increased Candidatus, Saccharibacteria and Firmicutes phyla abundance and reduced Fibrobacteres abundance At the genus level: Streptococcus abundance increased but not consistently across individual animals. Fecal pH and SCFA concentrations modified by diet but considerable inter-individual variation |
| Reference | Horses | Microbiota Analysis | Alpha-Diversity | Beta-Diversity | Composition |
|---|---|---|---|---|---|
| COLITIS and DIARRHEA | |||||
| [34] | 30 clinically healthy horses of two age groups (adult vs. geriatric) and 5 geriatric diarrheic horses | Fecal sample in rectum, V1-V2 region of the 16S rRNA gene | Not reported | Significant differences between healthy and diarrheic horses. Strong heterogeneity among the diarrheic horses | On average ↑ Proteobacteria and ↓ Fibrobacteres and Verrucomicrobia. Negative correlation between relative abundance of Verrucomicrobia and diarrhea score |
| [88] | Cecal and colonic tissues from 7 horses with acute diarrhea (post-mortem) of 3 horses free from digestive diseases (chronic arthritis and cervical stenosis) differing for sex, age, and breeds | V3-V4 region of the 16S rRNA | No difference colitis vs. control in both colon and caecum. In colitis horse, mucosa richness > content richness | Significant difference in mucosa and content in colon and caecum | Regardless of the intestinal compartment (colon or cecum) or the sampling site (luminal or mucosal), there were 27 taxa associated with healthy horses (LDA > 3) and 24 taxa associated with horses with colitis. ↑ Lactobacillus, Escherichia/Shigella, Enterobacteriaceae and Fusobacterium and ↓ Fibrobacter, Lachnospiraceae uncl., Clostridiales uncl., Fretibacterium and Bacteroidetes uncl. |
| [30] | 6 horses with chronic or acute colitis and 2 healthy donors to evaluate fecal microbial transplantation | Fecal swab and feces. V4 region of the 16S rRNA | ↓ richness between donor and diarrheic horses | No difference | ↑ Intestinimonas, unclassified Lactobacillales, Lactobacillus, and Streptococcus, when compared to the donors ↓ relative abundance of the genus Saccharofermentans |
| [27] | 10 healthy and 10 diarrheic horses differing for sex, age and breeds | Negative for C. difficile by feces culture. Rectal swabs for fecal collection, V1-V3 region of the 16S rDNA | ↓ richness and evenness; ↓ Shannon index | Significant difference | ↑ Actinobacillus, Porphyromonas, Roseburia ↓ RC9 gut group and Ruminococcaceae unclassified |
| [30] | 6 healthy horses and 10 colitis horses differing for sex, age and breeds | Negative cultures for Salmonella spp, as well as single negative fecal ELISA results for Clostridium perfringens enterotoxin and C. difficile toxins A and B. Fecal samples. V3-V5 Region of the 16S rRNA Gene | No significant differences in alpha-diversity | Significant difference | ↓ Actinobacteria and Spirochaetes; ↑ Fusobacteria among which F. necrophorum and F. nucleatum; ↓ Clostridia, Heliobacteriaceae, Lachnospiraceae, Eubacteriaceae, Peptococcaceae, Clostridiaceae and Ruminococcaceae. Among Clostridiaceae, ↓ Trepidimicrobium and Clostridium |
| COLIC | |||||
| [89] | 28 horses showing signs of colic into two study groups: horses with large intestinal colic (LC, n = 20) and horses with small intestinal colic (SC, n = 8). 24 clinically healthy adult horses. All horses were thoroughbreds | Fecal samples at D0 (admission) Amplicon sequencing of the V3-V4 region | ↓ number species observed and Shannon index in horses with large intestine colic | Significant difference between control, large colon and small intestine colic horses | Horses with small intestine colic: ↑ Firmicutes, ↓ Methanobacteriaceae and subdivision 5 Verrucomicrobia. LEfSe: ↑ Enterococcus, Lactobacillus, Acinetobacter, Bifidobacterium, Kurthia, Weissella, Rummeliibacillus, ↓ Methanobrevibacter, Coprococcus, Faecalitalea, Treponema, Akkermansia Horses with large intestine colic: ↑ Bacteroidetes, Lachnospiraceae, Streptococcaceae, Lactobacillaceae and Coriobacteriaceae, ↓ Verrucomicrobia. LEfSe: ↑ Enterococcus, Acinetobacter, Lactobacillus, E.coli/Shigella, Blautia, ↓ Methanobrevibacter, Unclassified bacteria Verrucomicrobia |
| [90] | 17 horses; 3 horses young, 8 mature and 6 as geriatric. 10 Thoroughbreds, 6 Warmbloods and 1 mixed-breed horses. Fourteen horses were admitted with a colic episode < 60 h and three horses were admitted with a history of colic ≥ 60 h. Different lesions of intestine | Fecal samples at D0 (admission), D1 and D3. Amplicon sequencing | ↓ number of species observed between D0 and D3; = D0 to D1; = D1 to D3 ↓ number of species observed and Shannon index in horses with colic ≥ 60 h | Significant difference depending on the time | ↓ Firmicutes from D0 to D1 and remained lower than admission on D3 Bacteroidetes and Proteobacteria ↑ in all horses, while Fibrobacteres ↓ from D0 to D3 in horses with colic ≥ 60 h. More profound changes in all horses with colic ≥ 60 h. |
| [91] | 9 horses with large intestinal forms of surgical colic and orthopaedic controls with general anaesthesia same initial antimicrobial and analgesic protocol than colic horses | Colonic and fecal samples at the admission D0, fecal every 2–3 days during hospitalization, weekly during the first month after hospital discharge and then every 2 weeks for a further 2 months. Amplicon sequencing V1-V2 regions | No significant differences in alpha-diversity of fecal microbiota between colic and control horses at admission | No significant differences in beta-diversity of fecal microbiota between colic and control horses at admission | ↑ 21 OTUs (mainly Fibrobacteres (n = 8), Bacteroidetes (n = 5) and Spirochaetes (n = 6)) ↓ 25 OTUs (Firmicutes (n = 9) and Bacteroidetes (n = 16)) in the fecal microbiota of case horses |
| [32] | Post-partum colic: 13 mares that developed colic, 13 mares that did not display colic and 5 nonpregnant controls | Fecal samples were collected approximately 14 D prior to the estimated foaling date, within 4 D after parturition, and 14 and 28 D after foaling. Episodes of colic were recorded. Amplicon sequencing of the V4 region | No significant differences in alpha-diversity of fecal microbiota neither in richness nor in evenness | Difference from 10 D before colic appearance | In the >10 D previous the colic: ↓ Firmicutes, ↑ Proteobacteria (↑ Rhodopseudomonas, uncl. Enterobacteriaceae and Enhydrobacter); <10 D before colic appearance: ↓ Firmicutes (↓ Sphingobacteriales, Acetovibrio, Ruminococcus), Bacteroidetes (uncl Bacteroidales) and Tenericutes. Firmicutes:Protebacteria relevance ratio Shorter time (<4 D before colic): ↓ Ruminococcaceae and Lachnospiraceae |
| FREE FECAL WATER (FFW) | |||||
| [92] | Case-control study with 100 healthy and 100 horses with FFW differing for sex, age and breeds | Fecal collection, 3 periods: Oct/Nov; Dec/Jan; Feb/March. Culture to determine the concentration of C. perfringens and C. difficile + V3-V4 regions of the 16S rRNA gene | No change in richness, evenness and in Shannon index | No change | Negative to C. perfringens and C.difficile. 14 genera differed in relative abundance between case and control horses within at least one sample collection. These genera belonged to the phylum Bacteroidetes (n = 2, ↑ Alloprevotella and 1 Bacteroidetes), Euryarchaeota (n = 1; ↓ Methanobrevibacter) and Firmicutes (n = 11; ↓ Bacillus, Solibacillus, Lachnoclostridium sensu stricto, Roseburia; ↑ Lactobacillus, Marvinbryanttia, Oribacterium, Ruminococcaceae UGC005, Saccharofermentans). Overall no big changes in fecal microbiota composition and diversity. |
| [93] | 10 horses with FFW for >12 months vs. 10 healthy horses differing for sex, age and breeds | Rectal collection of feces. V4 region of the 16S rRNA | No change in richness, evenness and in Shannon index | No change | No change |
| [94] | 16 horses with FFW; 15 healthy horses differing for sex, age and breeds | 1 fecal sample in spring, another in autumn. V4 region of the 16S rRNA gene | No change in richness, evenness and in Shannon index | No change | Differences in microbial community composition based on time point and health status were not observed on any taxonomic level. |
| Reference | Animal Characteristics | Microbiome Measures | Main Results vs. Healthy Horses |
|---|---|---|---|
| Laminitis | |||
| [33] | 10 normal horses and 8 horses with chronic laminitis | Feces. Amplicon sequencing, V5-V9 regions | ↑ number of OTUs and ↑ 2 OTUs of Clostridiales order Great inter-individual variability |
| [112] | 5 horses with OF-induced laminitis | Culture and molecular methods | ↑ complex Streptococcus bovis/equum, then of Lactobacillus sp. E. coli increased post-laminitis. |
| [118] | 20 horses, 8 control, 6 with acute laminitis induced by corn starch infusion, 6 with acute laminitis induced by OF infusion. | Feces. Amplicon sequencing, V4 region from cecum | No evaluation of alpha and beta-diversity ↑ Firmicutes, Lactobacillus, Streptococcus, Veillonella, Serratia ↓ Bacteroidetes, Bacteroidales, Bacillus and Solibacillus, Verrucomicrobia, Akkermansia, Ruminococcaceae and Veillonellaceae |
| [41] | 10 healthy horses, 6.7 y, 3 males, 7 mares; OF-induced laminitis | Feces. Amplicon sequencing, V4 region from feces Metabolomics from intestinal contents | ↓ fecal pH Alpha-diversity: ↓ Beta-diversity: ≠ ↓ Kiritimatiellaeota, Fibrobacteres, Tenericutes, Lentisphaerae, Elusimicrobiae, Verrucomicrobia, Planctomycetes ↑ Lactobacillus, Megasphaera, Allisonella ↓ Fibrobacter, Phascolarctobacterium, Papillibacter, Alloprevotella, Candidatus soleaferrea, Oribacterium, Akkermansia, Elusimicrobium Biomarkers (Lefse): Lactobacillus (L. gasseri and L. delbrueckii), Megasphaera (M. elsdenii), Sharpea, Streptococcus, Prevotella-sp_DJF_CP65 Metabolites: ≠ clusters, 53 and 83 metabolites with higher and lower concentrations respectively. Enrichment of ABC transporters, glycerophospholipid metabolism, inflammatory mediator of TRP channels, lysine degradation, vitamin digestion and absorption, tyrosine metabolism Correlation network: asparagine and 9-hydroxy-10E, 12Z-octadecadienoic acid correlated + with Lactobacillus and Megasphaera |
| EMS and insulin dysregulation | |||
| [119] | 16 mixed-breed ponies classified according to their insulin dysregulation (5 healthy (NID), 11 medium (MID) to severe insulin-dysregulated (SID)) subjected to a dietary change: adding pasture to a hay diet | Feces. Amplicon sequencing V3-V4 regions, feces | Alpha-diversity: = except evenness, lower in MID ponies than in NID and SID. Beta-diversity: = ↑ Firmicutes and Bacteroidetes in MID (with higher blood GLP-1 concentration) ↓ Christensenellaceae R-7 group, ↑ Rikenellaceae and Kiritimatiellae in MID vs. NID |
| [113] | 20 horses, mixed breeds and genders, 10 EMS; 10 non-EMS horses based on insulin dysregulation estimated through OST, general/regional adiposity and a history/predisposition to laminitis. Natural EMS | Feces. Amplicon sequencing, V4 region PICRUSt functional inference | Alpha-diversity: = Beta-diversity: ≠ PICRUSt data: = 12 significant OTUs (LEfSe): ↑ Verrucomicrobia subdivision 5 (now Kiritimatiellaeota), Cellulosilyticum, Elusimicrobium, Clostridium cluster XI and Lactobacillus, in EMS group; and ↓ Fibrobacter, Uncl. Lachnospiraceae, Anaerovorax, Uncl. Rhodospiracellaceae, Uncl. Flavobacteriaceae, Saccharofermentans, Ruminococcus |
| Obese/Weight loss management | |||
| [18] | 35 Welsh-section A ponies mares, 11 aged, 12 control and 12 obese. Glucose-insulin tolerance, digestibility evaluated in each phenotypic group. Controlled feeding, hay ration | Feces. Amplicon sequencing, V1-V2 regions, SCFAs, pH Metabolome (FT-IR spectroscopy) | Same copy numbers of bacteria, fungi and protozoa ↑ Shannon and Simpson index Beta-diversity: ≠ ↑ Bacteroidetes, Firmicutes and Actinobacteria, ↓ Fibrobacter, ↑ Pseudoflavonifractor ↑ fecal pH SCFAs: =, no change in fecal metabolome |
| [107] | 78 horses: 24 lean, 17 normal and 37 obese | Feces. Amplicon sequencing, V4-V5 regions. Network construction between microbial OTUs and blood analytes | ↑ Shannon index, chao1, observed OTUs and phylogenetic diversity Phyla level: = but ↑ Firmicutes: Bacteroidetes ratio in obese. 24 OTUs with different relative abundance between normal and obese horses BCS Obese positively correlated with Actinobacteria, Firmicutes (Ruminococcaceae and Lachnospiraceae) and Bacteroidetes Network: obese BCS positively associated Campylobacter sp., Collinsella sp., Prevotellaceae, Selenomonas sp., Blautia sp., Mogibacterium sp., Adlercreutzi sp., Erysipelotrichaceae, Propinibacteriaceae, Butyrivibrio sp., Ruminococcaceae, Sutterella sp. |
| [120] | 10 Shetland ponies and 10 warmblood horses subjected to a 2-y body weight gain program | Feces. Amplicon sequencing, V3-V4 regions, SCFAs, lactate | Ponies: ↓ number of OTUs ↑ Firmicutes and Actinobacteria ↓ lactate Horses: ↓ Fibrobacteres, ↑ Actinobacteria; ↓ Ruminococcaceae, ↑ Lachnospiraceae ↓ isobutyrate and lactate |
| [121] | 12 mature obese horses and ponies, mixed breed. 2 restricted diets for 16 w. (traditional concentrate + hay vs. nutrient balancer (hay only)) | Feces. Amplicon sequencing, V1-V2 regions, 2 time points: 10 and 16 w. | Alpha-diversity: = Absence of Fibrobacteres at the start of the study Weight loss: ↑ Anaeroplasma, several unclassified Firmicutes and Bacteroidetes, Anaerophaga, Phocaeicola and ↓ Lactobacillus, Streptococcus, Butyrivibrio, Roseburia and uncl. Acidaminococcaceae |
| [122] | 20 obese and 20 normal horses matched by farm origin. BCS >= 7: obese, 3 < BCS < 7 | Feces. Amplicon sequencing, V4 region. Fecal metabolome and serum lipidome | Alpha-diversity: = Beta-diversity: = 8 OTUs ≠ among which ↓ Methanobrevibacter, Ruminococcus, uncl. Lachnospiraceae, uncl. Bacteroidetes (2 OTUs) 57 metabolites ≠: 18 higher and 39 lower in obese horses. ↑ isocitrate, citrate, aconitate TCA cycle, ↓ vit E metabolism 146 lipids affected, 110 higher and 36 lower in obese horses: ↑ free fatty acids 14:0, 16:1, 18:1, 18:2, 18:3 and of cholesteryl esters, diacylglycerols, phosphatidylcholines |
| [123] | 14 overweighed horses and ponies, 2 groups: control and fed with a restricted diet (2 vs. 1.4% BW DMI), 6 weeks | Feces. Amplicon sequencing, V3-V4 regions. Fecal metabolome | Weight loss program: Alpha-diversity: ↑ Beta-diversity: ≠ ↓ Eubacteriaceae, Pseudomonaceae, ↑ Coprococcus and class of Clostridia in the treated group. No change in metabolome. Network: positive correlation between Rikenellaceae and urocanic acid, Ruminoccocaceae with propionate, and Ruminoccocaceae and Phascolarctobacterium with urocanic acid. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaucheyras-Durand, F.; Sacy, A.; Karges, K.; Apper, E. Gastro-Intestinal Microbiota in Equines and Its Role in Health and Disease: The Black Box Opens. Microorganisms 2022, 10, 2517. https://doi.org/10.3390/microorganisms10122517
Chaucheyras-Durand F, Sacy A, Karges K, Apper E. Gastro-Intestinal Microbiota in Equines and Its Role in Health and Disease: The Black Box Opens. Microorganisms. 2022; 10(12):2517. https://doi.org/10.3390/microorganisms10122517
Chicago/Turabian StyleChaucheyras-Durand, Frédérique, Audrey Sacy, Kip Karges, and Emmanuelle Apper. 2022. "Gastro-Intestinal Microbiota in Equines and Its Role in Health and Disease: The Black Box Opens" Microorganisms 10, no. 12: 2517. https://doi.org/10.3390/microorganisms10122517