
Genomic and Phylogenetic Characterization of Rhodopseudomonas infernalis sp. nov., Isolated from the Hell Creek Watershed (Nebraska)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Isolation and Cultivation
2.2. DNA Purification
2.3. Next Generation Sequencing
2.4. Whole Genome Comparison
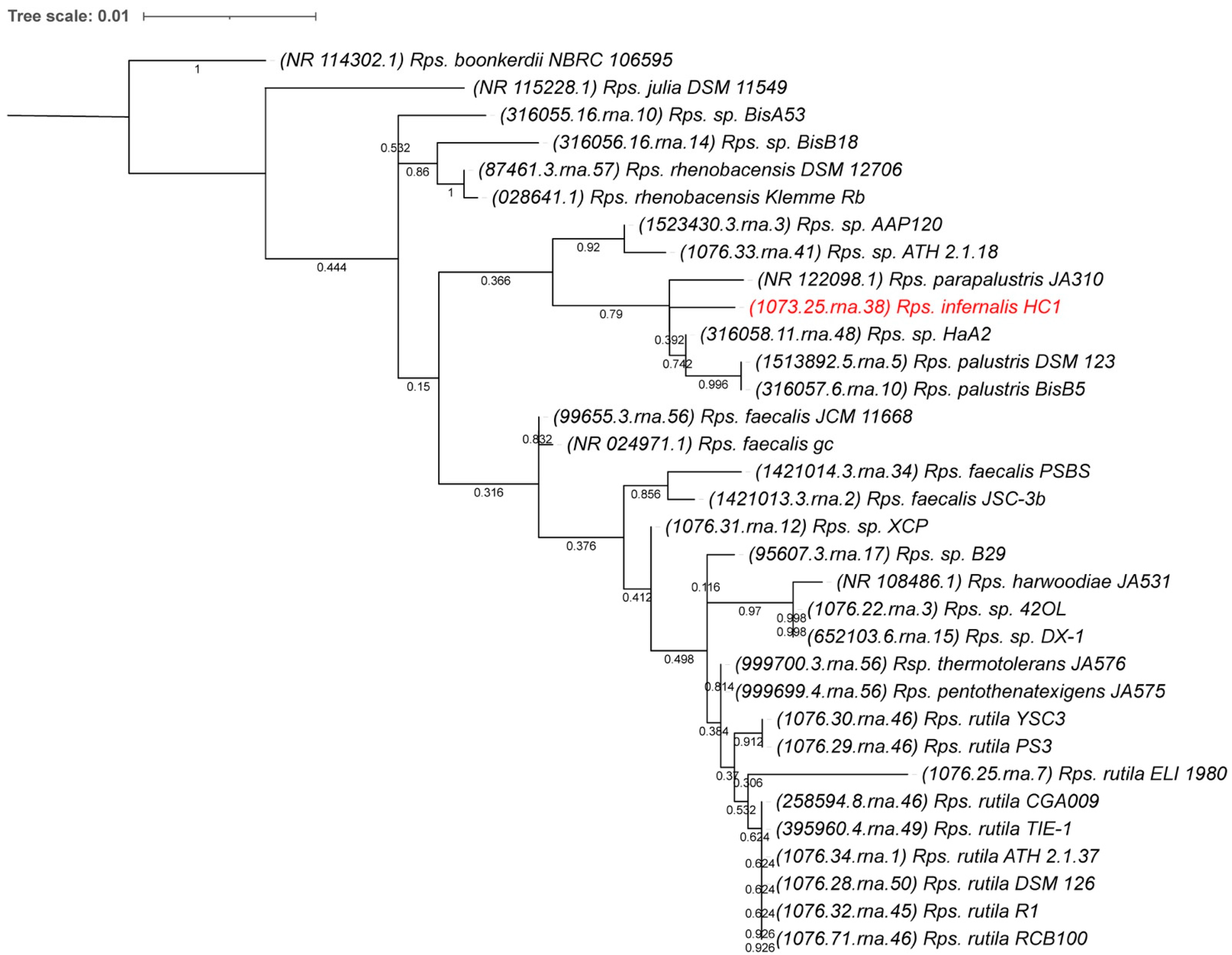
2.5. 16S rDNA Phylogenetic Comparison
3. Results and Discussion
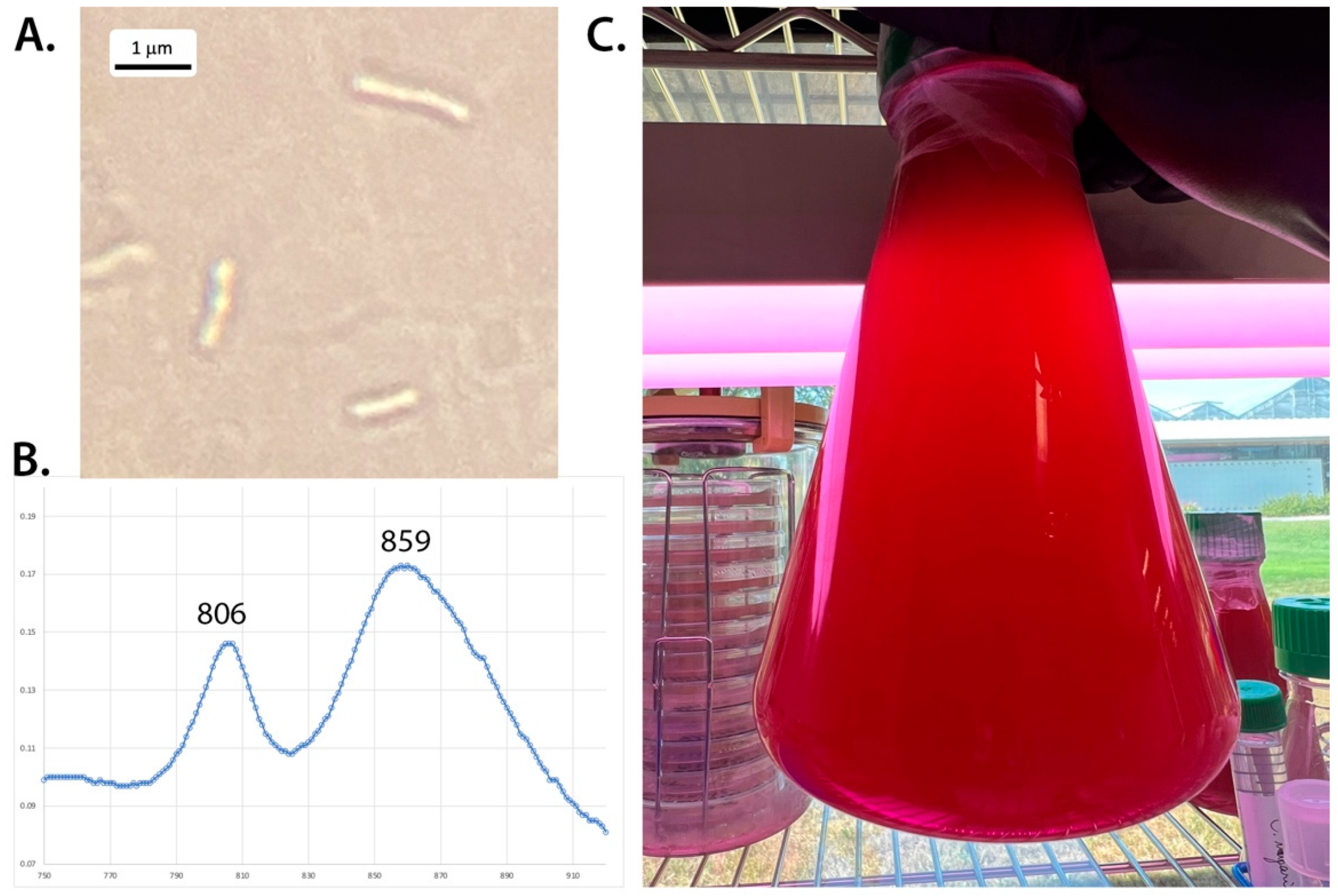
3.1. Morphology and Photopigments
3.2. Physiological Characteristics
3.3. Genome Sequence
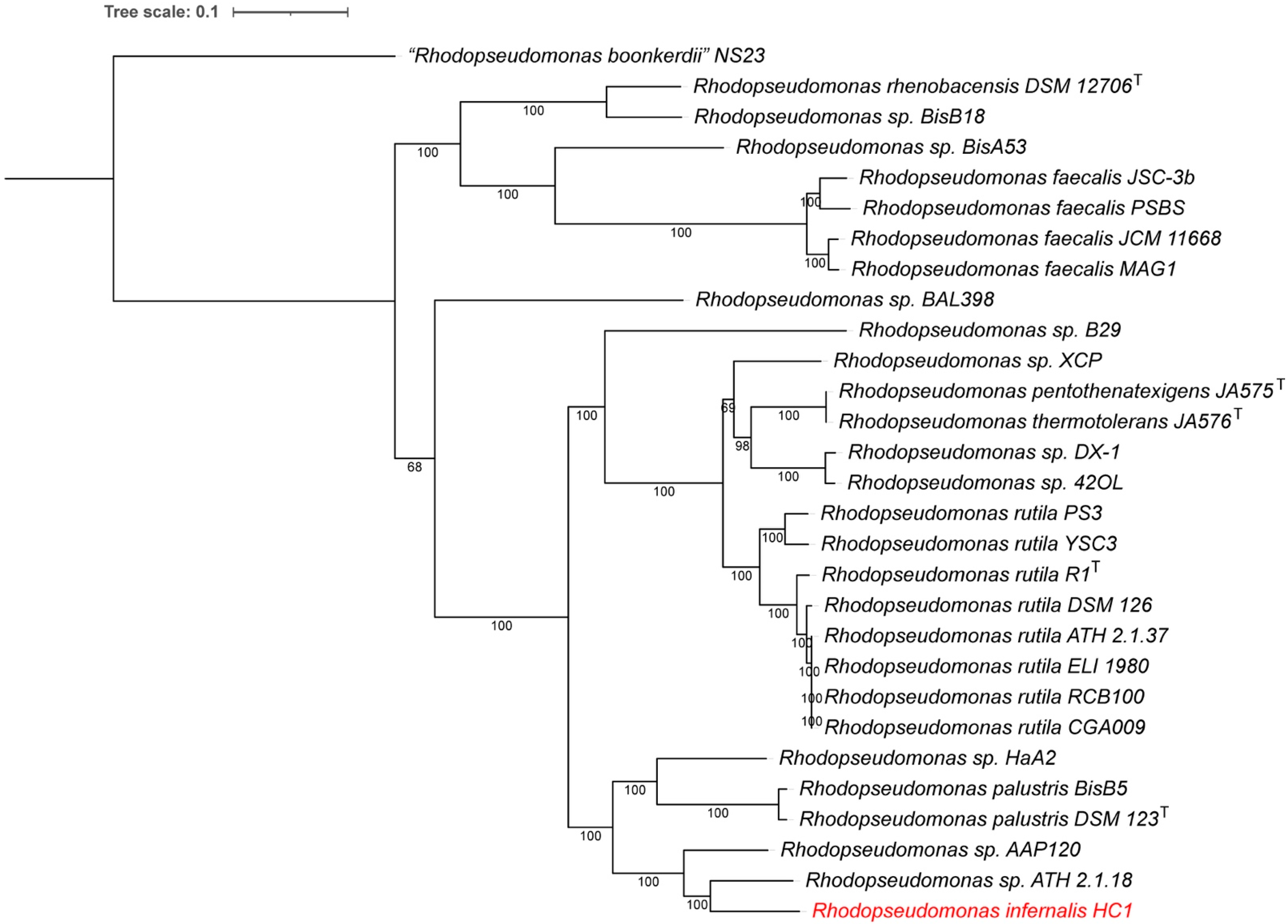
3.4. Whole Genome-Based Phylogenetic Analysis
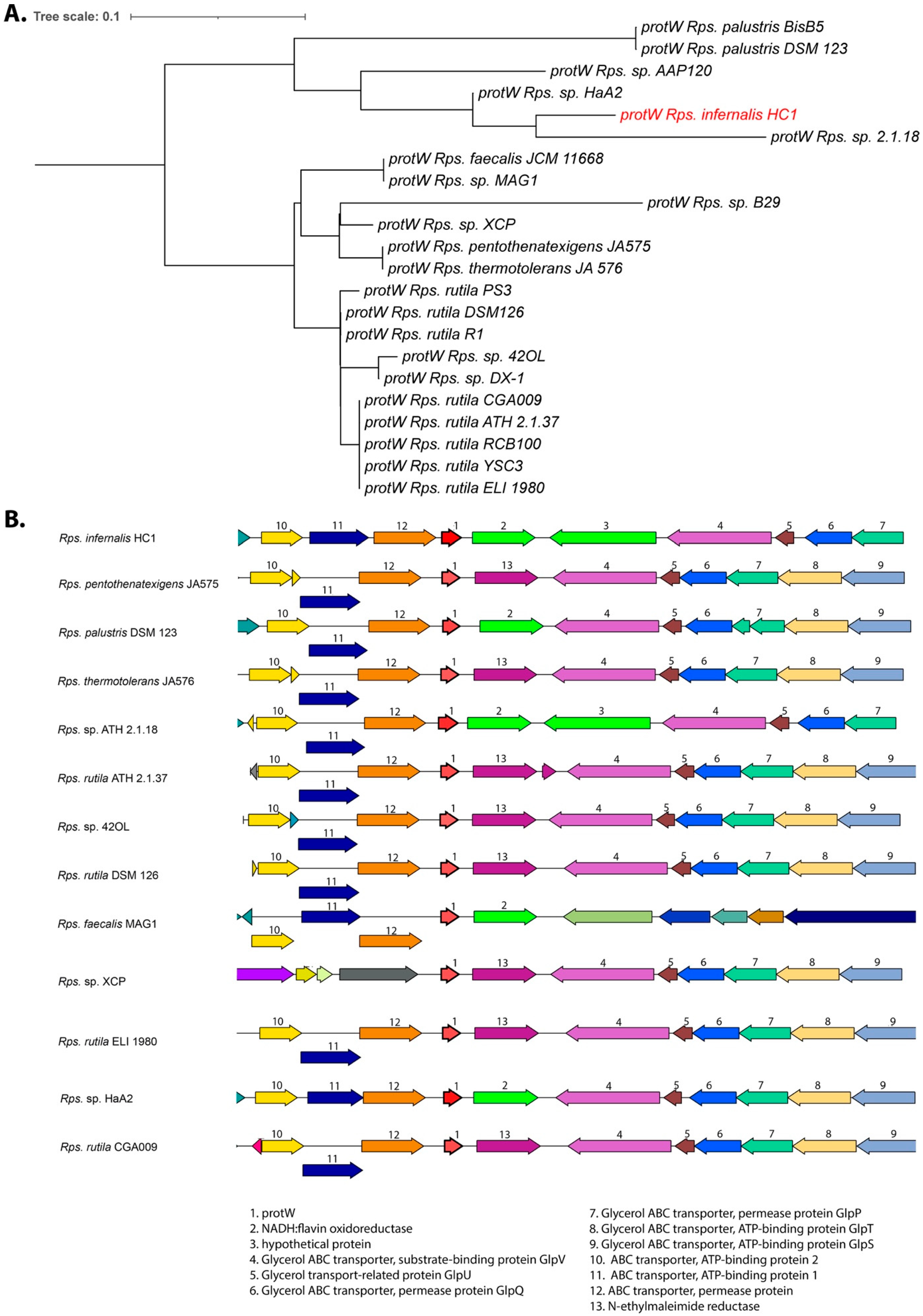
3.5. Photosynthetic Gene Cluster
3.6. Growth and Respiration Genes
3.7. Nitrogen Fixation and Hydrogen Production
3.8. Aromatic Compound Degradation
3.9. Antibiotic Resistance
3.10. Description of Rhodopseudomonas infernalis sp. nov.
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kyndt, J.A. Microbial diversity of the Hell Creek watershed at the Tri-Faith Community in Nebraska, based on 16S rRNA gene amplicon sequencing. Microbiol. Res. Announc. 2020, 9, e00467-20. [Google Scholar] [CrossRef]
- Oda, Y.; Wanders, W.; Huisman, L.A.; Meijer, W.G.; Gottschal, J.C.; Forney, L.J. Genotypic and phenotypic diversity within species of purple nonsulfur bacteria isolated from aquatic sediments. Appl. Environ. Microbiol. 2002, 68, 3467–3477. [Google Scholar] [CrossRef] [Green Version]
- Rayyan, A.; Meyer, T.; Kyndt, J. Draft whole-genome sequence of the purple photosynthetic bacterium Rhodopseudomonas palustris XCP. Microbiol. Res. Announc. 2018, 7, e00855-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraishi, A.; Kitamura, H. Distribution of phototropic purple nonsulfur bacteria in activated sludge systems and other aquatic environments. Bull. Jpn. Soc. Sci. Fish 1984, 50, 1929–1937. [Google Scholar] [CrossRef]
- Venkidusamy, K.; Megharaj, M. A novel electrophototrophic bacterium Rhodopseudomonas palustris strain RP2 exhibits hydrocarbonoclastic potential in anaerobic environments. Front. Microbiol. 2016, 7, 1071. [Google Scholar] [CrossRef] [Green Version]
- Akiba, T.; Usami, R.; Horikoshi, K. Rhodopseudomonas rutila, a new species of nonsulfur purple photosynthetic bacteria. Int. J. Syst. Bacteriol. 1983, 33, 551–556. [Google Scholar] [CrossRef]
- Madigan, M.T.; Gest, H. Selective enrichment and isolation of Rhodopseudomonas palustris using trans-cinnamic acid as sole carbon source. FEMS Microbiol. Ecol. 1988, 53, 53–58. [Google Scholar] [CrossRef]
- Cetinkaya Donmez, G.; Ozturk, A.; Cakmakci, L. Properties of the Rhodopseudomonas palutris strains isolated from an alkaline lake in Turkey. Turk. J. Biol. 1999, 23, 457–464. [Google Scholar]
- Kim, M.K.; Choi, K.M.; Yin, C.R.; Lee, K.Y.; Im, W.T.; Lim, J.H.; Lee, S.T. Odorous swine wastewater treatment by purple non-sulfur bacteria, Rhodopseudomonas palustris, isolated from eutrophicated ponds. Biotechnol. Lett. 2004, 26, 819–822. [Google Scholar] [CrossRef]
- Imhoff, J.F. Genus Rhodopseudomonas. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Springer: New York, NY, USA, 2005; Volume 2, pp. 473–476. [Google Scholar]
- Larimer, F.; Chain, P.; Hauser, L.; Lamerdin, J.; Malfatti, S.; Do, L.; Land, M.; Pelletier, D.; Beatty, J.T.; Lang, A.; et al. Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat. Biotechnol. 2004, 22, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, B.; Wilkins, M.; Saha, R. Rhodopseudomonas palustris: A biotechnology chassis. Biotechnol. Adv. 2022, 60, 108001. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Larimer, F.W.; Chain, P.S.; Malfatti, S.; Shin, M.V.; Vergez, L.M.; Hauser, L.; Land, M.L.; Braatsch, S.; Beatty, J.T.; et al. Multiple genome sequences reveal adaptations of a phototrophic bacterium to sediment microenvironments. Proc. Natl. Acad. Sci. USA 2008, 105, 18543–18548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraud, E.; Zappa, S.; Vuillet, L.; Adriano, J.M.; Hannibal, L.; Fardoux, J.; Berthomieu, C.; Bouyer, P.; Pignol, D.; Verméglio, A. A new type of bacteriophytochrome acts in tandem with a classical bacteriophytochrome to control the antennae synthesis in Rhodopseudomonas palustris. J. Biol. Chem. 2005, 280, 32389–32397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harwood, C.S. Rhodopseudomonas palustris. Trends Microbiol. 2022, 30, 307–308. [Google Scholar] [CrossRef]
- Evans, K.; Georgiou, T.; Hilton, T.; Fordham-Skelton, A.P.; Papiz, M.Z. Bacteriophytochromes control photosynthesis in Rhodopseudomonas palustris. In The Purple Phototrophic Bacteria; Hunter, C.N., Daldal, F., Thurnauer, M.C., Beatty, J.T., Eds.; Springer: Heidelberg, The Netherlands, 2009; pp. 799–809. [Google Scholar]
- Harwood, C.S.; Gibson, J. Anaerobic and aerobic metabolism of diverse aromatic compounds by the photosynthetic bacterium Rhodopseudomonas palustris. Appl. Environ. Microbiol. 1988, 54, 712–717. [Google Scholar] [CrossRef] [Green Version]
- McGrath, J.E.; Hartfoot, C.G. Reductive dehalogenation of halocarboxylic acids by the phototrophic genera Rhodospirillum and Rhodopseudomonas. Appl. Environ. Microbiol. 1997, 63, 3333–3335. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Luo, X.; Cheng, J.; Peng, J.; Zhang, D.; Lui, Y. Genome sequence of pyrethroid-degrading bacterium Rhodopseudomonas palustris strain JSC-3b. Genome Announc. 2014, 2, e01228-13. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.W.; Seol, E.H.; Kim, M.S.; Park, S. Photoproduction of hydrogen from acetate by a chemoheterotrophic bacterium Rhodopseudomonas palustris P4. Int. J. Hydrogen Energy 2004, 29, 1115–1121. [Google Scholar] [CrossRef]
- Carlozzi, P. Hydrogen photoproduction by Rhodopseudomonas palustris 42OL cultured at high irradiance under semicontinuous regime. J. Biomed. Biotechnol. 2012, 2012, 590693. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, C.B. The culture, general physiology, morphology, and classification of the non-sulfur purple and brown bacteria. Bacteriol. Rev. 1944, 8, 1–118. [Google Scholar] [CrossRef]
- Hougardy, A.; Tindall, B.J.; Klemme, J.-H. Rhodopseudomonas rhenobacensis sp. nov., an new nitrate-reducing purple non-sulfur bacterium. Int. J. Syst. Evol. Microbiol. 2000, 50, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Hiraishi, A.; Okamura, K. Rhodopseudomonas telluris sp. nov., a phototrophic alphaproteobacterium isolated from paddy soil. Int. J. Syst. Evol. Microbiol. 2017, 67, 3369–3374. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F.; Meyer, T.E.; Kyndt, J.A. Genomic and genetic sequence information of strains assigned to the genus Rhodopseudomonas reveal the great heterogeneity of the group and identify strain Rhodopseudomonas palustris DSM 123T as an authentic type strain of this species. Int. J. Syst. Evol. Microbiol. 2020, 70, 3932–3938. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.; Rayyan, A.; Meyer, T.; Kyndt, J. Draft whole-genome sequence of the purple nonsulfur photosynthetic bacterium Rhodopseudomonas rutila R1. Microbiol. Resour. Announc. 2018, 7, e01267-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, P.; Wall, J.D.; Gest, H. Characterization of Rhodopseudomonas capsulata. Arch. Microbiol. 1975, 105, 207–216. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Fromsma, K.; Gerdes, S.; Glass, E.M.; Kabul, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R.; Glöckner, F.O.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 16, btv681. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.J.B. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating the human-ape split by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiedzki, D.M.; Dilbeck, P.L.; Tang, Q.; Mothersole, D.J.; Martin, E.C.; Bocian, D.F.; Holten, D.; Hunter, C.N. Functional characteristics of spirilloxanthin and keto-bearing analogues in light-harvesting LH2 complexes from Rhodobacter sphaeroides with a genetically modified carotenoid synthesis pathway. Biochim. Et Biophys. Acta (BBA)–Bioenerg. 2015, 1847, 640–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzziotti, D.; Adessi, A.; Faraloni, C.; Torzillo, G.; De Philippis, R. Acclimation strategy of Rhodopseudomonas palustris to high light irradiance. Microbiol. Res. 2017, 197, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E.; Ebers, J. Taxonomic parameters revisited: Tarnished gold standards. Microbiol. Today 2006, 33, 152–155. [Google Scholar]
- Braatsch, S.; Bernstein, J.R.; Lessner, F.; Morgan, J.; Liao, J.C.; Harwood, C.S.; Beatty, J.T. Rhodopseudomonas palustris CGA009 has two functional ppsR genes, each of which encodes a repressor of photosynthesis gene expression. Biochemistry 2006, 45, 14441–14451. [Google Scholar] [CrossRef]
- Giraud, E.; Fardoux, J.; Fourrier, N.; Hannibal, L.; Genty, B.; Bouyer, P.; Dreyfus, B.; Verméglio, A. Bacteriophytochrome controls photosystem synthesis in anoxygenic bacteria. Nature 2002, 417, 202–205. [Google Scholar] [CrossRef]
- Robertson, S.L.; Meyer, T.E.; Kyndt, J.A. Draft genome sequence of Rhodopseudomonas palustris 2.1.18. Genbank, 2018; unpublished. Available online: https://www.ncbi.nlm.nih.gov/nuccore/QYYD00000000.1(accessed on 12 October 2022).
- Swainsbury, D.J.K.; Qian, P.; Jackson, P.J.; Faries, K.M.; Niedzwiedzki, D.M.; Martin, E.C.; Farmer, D.A.; Malone, L.A.; Thompson, R.F.; Ranson, N.A.; et al. Structures of Rhodopseudomonas palustris RC-LH1 complexes with open or closed quinone channels. Sci. Adv. 2021, 7, eabe2631. [Google Scholar] [CrossRef]
- Badger, M.R.; Bek, E.J. Multiple Rubisco forms in proteobacteria: Their functional significance in relation to CO2 acquisition by the CBB cycle. J. Exp. Bot. 2008, 59, 1525–1541. [Google Scholar] [CrossRef] [Green Version]
- Kyndt, J.A.; Aviles, F.A.; Imhoff, J.F.; Künzel, S.; Neulinger, S.C.; Meyer, T.E. Comparative genome analysis of the photosynthetic Betaproteobacteria of the genus Rhodocyclus: Heterogeneity within strains assigned to Rhodocyclus tenuis and description of Rhodocyclus gracilis sp. nov. as a new species. Microorganisms 2022, 10, 649. [Google Scholar] [CrossRef]
- Sucharitakul, J.; Tinikul, R.; Chaiyen, P. Mechanisms of reduced flavin transfer in the two-component flavin-dependent monooxygenases. Arch. Biochem. Biophys. 2014, 555–556, 33–46. [Google Scholar] [CrossRef]
- Tsai, S.C.; Li, Y.K. Purification and characterization of a catechol 1,2-dioxygenase from a phenol degrading Candida albicans TL3. Arch. Microbiol. 2007, 187, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, S.H.; Ryu, S.J.; Kang, C.S.; Suma, Y.; Kim, H.S. Effective biochemical decomposition of chlorinated aromatic hydrocarbons with a biocatalyst immobilized on a natural enzyme support. Bioresour. Technol. 2013, 141, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Eudes, A.; Berthomieu, R.; Hao, Z.; Zhao, N.; Benites, V.T.; Baidoo, E.E.K.; Loqué, D. Production of muconic acid in plants. Metab. Eng. 2018, 46, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Bromfield, E.S.P.; Cloutier, S.; Tambong, J.T.; Thi, T.V.T. Soybeans inoculated with root zone soils of Canadian native legumes harbour diverse and novel Bradyrhizobium spp. that possess agricultural potential. Syst. Appl. Microbiol. 2017, 40, 440–447. [Google Scholar] [CrossRef]
- Singer, A.C.; Shaw, H.; Rhodes, V.; Hart, A. Review of antimicrobial resistance in the environment and its relevance to environmental regulators. Front. Microbiol. 2016, 7, 1728. [Google Scholar] [CrossRef] [Green Version]
- Kraemer, S.A.; Ramachandran, A.; Perron, G.G. Antibiotic pollution in the environment: From microbial ecology to public policy. Microorganisms 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, D.G.J.; Flach, C.F. Antibiotic resistance in the environment. Nat. Rev. Microbiol. 2022, 20, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Welander, P.V.; Doughty, D.M.; Wu, C.-H.; Mehay, S.; Summons, R.E.; Newman, D.K. Identification and characterization of Rhodopseudomonas palustris TIE-1 hopanoid biosynthesis mutants. Geobiology 2012, 10, 163–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Chai, C.; Shao, L.; Yao, J.; Wang, Y. Metabolic engineering of Rhodopseudomonas palustris for squalene production. J. Ind. Microbiol. Biotechnol. 2016, 43, 719–725. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Strain HC1 | Rps. palustris DSM123 | Rps. julia DSM11549 | Rps. rhenobacensis Rb | |
|---|---|---|---|---|---|
| cell shape | Rod | Rod | Rod | Rod | |
| Cell width (um) | 0.2–0.5 | 0.8–1.2 | 1.0–1.5 | 0.5–0.8 | |
| Cell length (um) | 1.2–2.0 | 2.0–3.0 | 2.5 | 1.5–2.0 | |
| Motility | + | + | + | + | |
| pH optimum | 7–7.5 | 6–9 | 6–6.5 | 5–8 | |
| G+C content (mol%) | 65.3 * | 64.1 * | 63.5 | 65.4 | |
| Abs. maxima near IR (nm) | 806, 859 | 800, 861–863 | 803, 850 | 805, 878 | |
| Organic carbon sources: | |||||
| Propionate | - | + | + | - | |
| Citrate | - | (+) | - | - | |
| Malate | + | + | + | + | |
| Tartrate | + | - | - | + | |
| D-Gluconate | + | + | nd | + | |
| L-Aspartate | + | - | + | nd | |
| L-Glutamate | + | - | - | - | |
| Casamino acids | + | + | nd | nd | |
| L-Arginine | + | - | nd | - | |
| Benzoate | - | + | - | - | |
| D-Glucose | (+) | (+) | + | - | |
| D-Fructose | - | (+) | + | - | |
| Fumarate | + | + | + | + | |
| Isolation source | Pond water | Lake sediment | Acidic sulfide spring | Eutrophic pond | |
| Rps rutila DSM 126 | |||||||||||||||||||
| 97.6 | Rps rutila TIE-1 | ||||||||||||||||||
| 97.3 | 97.3 | Rps rutila R1T | |||||||||||||||||
| 88.3 | 88.2 | 88.3 | Rps pantothenatexigens JA575T | ||||||||||||||||
| 88.3 | 88.2 | 88.2 | 100 | Rps thermotolerans JA576T | |||||||||||||||
| 88.4 | 88.3 | 88.4 | 89.6 | 89.6 | Rps sp. DX1 | ||||||||||||||
| 88.1 | 88 | 88.1 | 89.3 | 89.3 | 88 | Rps sp. XCP | |||||||||||||
| 82 | 82.8 | 82.1 | 82.3 | 82.3 | 82.1 | 82.1 | Rps sp. AAP120 | ||||||||||||
| 81.6 | 81.5 | 81.6 | 82.1 | 82.1 | 81.8 | 81.6 | 87.1 | Rps sp. ATH 2.1.18 | |||||||||||
| 81.7 | 81.6 | 81.6 | 82 | 82 | 81.7 | 81.7 | 86.9 | 87.9 | Rps infernalis HC1 | ||||||||||
| 81.8 | 81.7 | 81.8 | 82.3 | 82.4 | 82.3 | 81.8 | 84 | 83.9 | 83.8 | Rps sp. HaA2 | |||||||||
| 81.2 | 81.1 | 81.2 | 81.6 | 81.6 | 81.3 | 81.2 | 83.3 | 82.9 | 83.1 | 85.6 | Rps palustris DSM 123T | ||||||||
| 81.4 | 81.6 | 81.4 | 81.8 | 81.8 | 81.5 | 81.1 | 83.6 | 83 | 83.1 | 85.7 | 98.2 | Rps palustrus BisB5 | |||||||
| 81 | 81 | 81.1 | 81.5 | 81.5 | 81.1 | 81.3 | 81.9 | 81.6 | 81.3 | 81.1 | 80.5 | 80.4 | Rps sp. B29 | ||||||
| 78.4 | 78.2 | 78.4 | 78.7 | 78.7 | 78.6 | 78.2 | 78.5 | 78.5 | 78.4 | 79.2 | 79.2 | 79.4 | 77.9 | Rps sp. BisA53 | |||||
| 77.9 | 77.8 | 77.8 | 78.3 | 78.2 | 78 | 77.7 | 77.8 | 77.8 | 77.3 | 78.3 | 77.9 | 78.2 | 77.5 | 80.9 | Rps faecalis JCM 11668T | ||||
| 78.6 | 78.8 | 78.7 | 78.9 | 78.9 | 78.6 | 78.6 | 78.9 | 78.9 | 78.6 | 79.6 | 79.4 | 79.5 | 78.5 | 81.2 | 79.3 | Rps. rhenobacensis DSM 12706 | |||
| 78.2 | 78.1 | 78.2 | 78.5 | 78.5 | 78.4 | 77.9 | 78.3 | 78.4 | 78.1 | 79.2 | 79 | 79.2 | 77.9 | 81.4 | 79.3 | 89.3 | Rps sp. BisB18 | ||
| 78.6 | 78.5 | 78.6 | 79 | 79 | 78.7 | 78.7 | 79.1 | 78.8 | 78.7 | 80 | 79.4 | 79.7 | 78.2 | 79.5 | 77.8 | 80.5 | 80.7 | Rps sp. BAL398 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Humphrey, C.E.; Burnett, N.; Dubey, S.; Kyndt, J.A. Genomic and Phylogenetic Characterization of Rhodopseudomonas infernalis sp. nov., Isolated from the Hell Creek Watershed (Nebraska). Microorganisms 2022, 10, 2024. https://doi.org/10.3390/microorganisms10102024
Humphrey CE, Burnett N, Dubey S, Kyndt JA. Genomic and Phylogenetic Characterization of Rhodopseudomonas infernalis sp. nov., Isolated from the Hell Creek Watershed (Nebraska). Microorganisms. 2022; 10(10):2024. https://doi.org/10.3390/microorganisms10102024
Chicago/Turabian StyleHumphrey, Christine E., Nicole Burnett, Shivangi Dubey, and John A. Kyndt. 2022. "Genomic and Phylogenetic Characterization of Rhodopseudomonas infernalis sp. nov., Isolated from the Hell Creek Watershed (Nebraska)" Microorganisms 10, no. 10: 2024. https://doi.org/10.3390/microorganisms10102024