

The Promises, Challenges, and Opportunities of Omics for Studying the Plant Holobiont
 , ,
, , 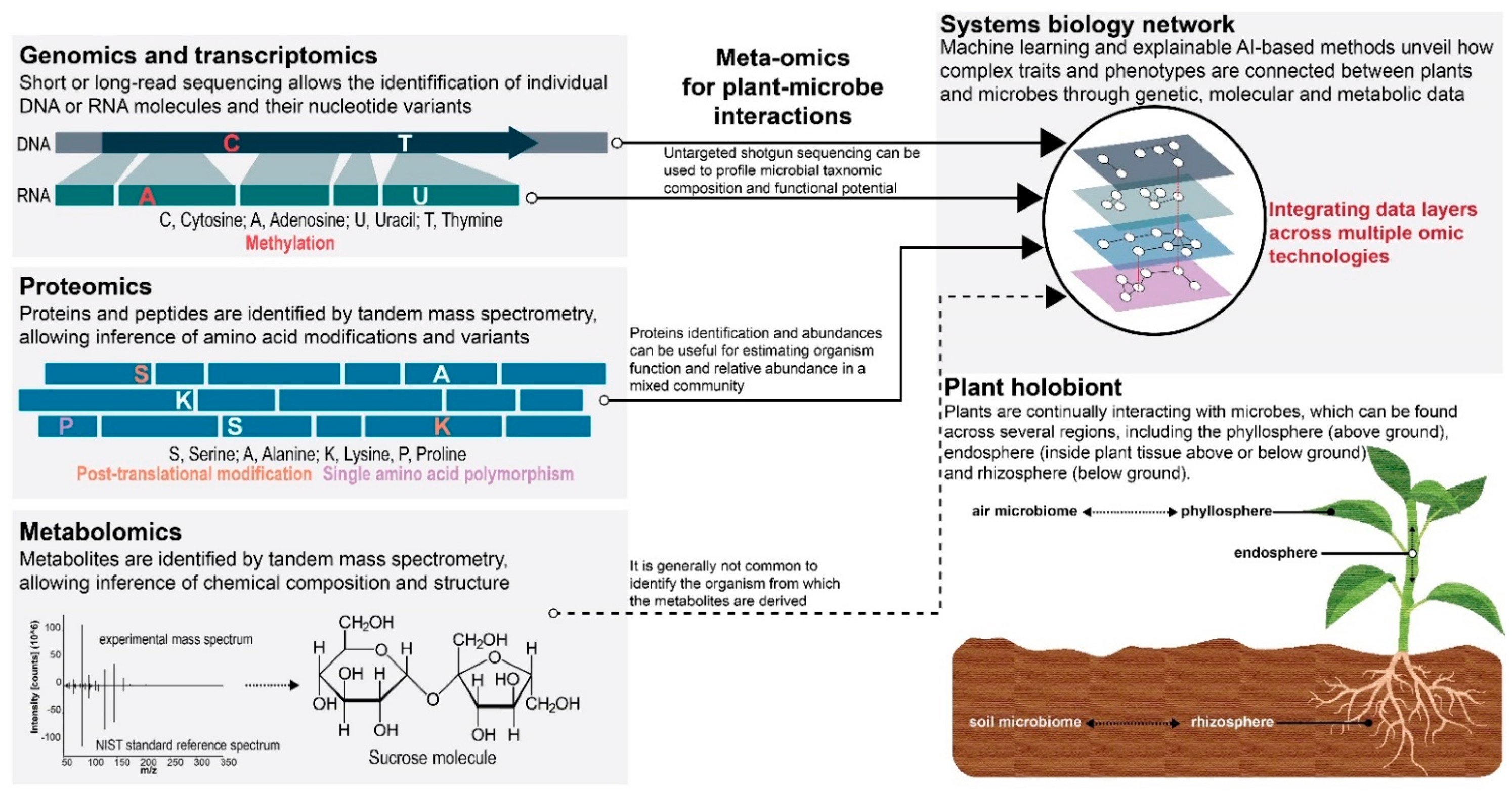{kind=link}
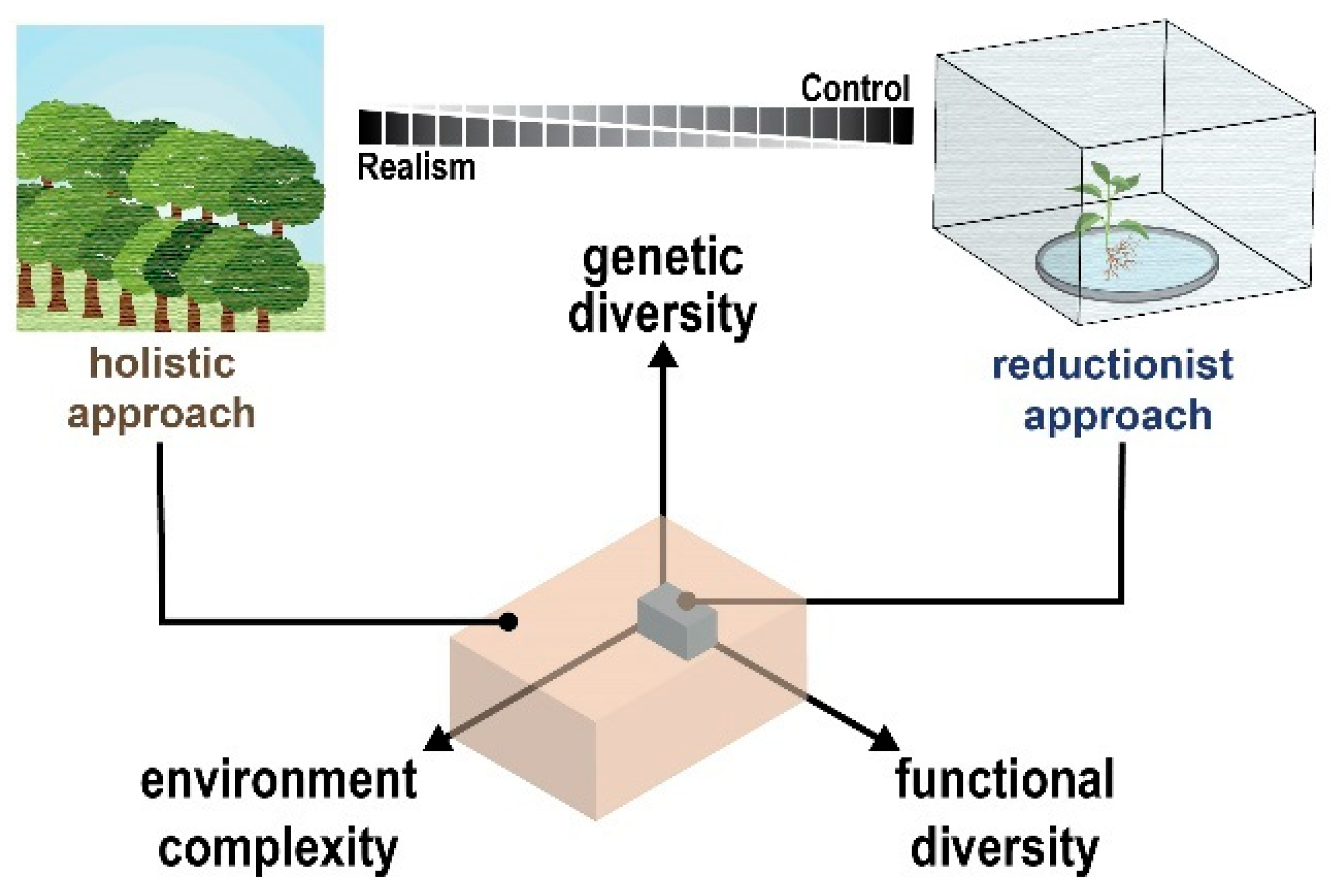{kind=link}
Abstract
:1. Introduction
2. Advancements in Omics Are Key to Defining Plant Holobionts
3. From Genes to Ecosystems: Studying Plant–Microbe Interactions across the Complexity Landscape
3.1. Recent Advancements and Current Impediments for Genomics, Transcriptomics, Proteomics, and Metabolomics for Studying the Plant Holobiont
3.1.1. Genomic and Transcriptomics
3.1.2. Proteomics
3.1.3. Metabolomics
3.1.4. Integrative Systems Biology
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dai, X.; Shen, L. Advances and Trends in Omics Technology Development. Front. Med. 2022, 9, 911861. [Google Scholar] [CrossRef]
- Gamalero, E.; Bona, E.; Glick, B.R. Current Techniques to Study Beneficial Plant-Microbe Interactions. Microorganisms 2022, 10, 1380. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant-microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Baedke, J.; Fabregas-Tejeda, A.; Nieves Delgado, A. The holobiont concept before Margulis. J. Exp. Zool. B Mol. Dev. Evol. 2020, 334, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Lyu, D.; Zajonc, J.; Page, A.; Tanney, C.A.S.; Shah, A.; Monjezi, N.; Msimbira, L.A.; Antar, M.; Nazari, M.; Backer, R.; et al. Plant Holobiont Theory: The Phytomicrobiome Plays a Central Role in Evolution and Success. Microorganisms 2021, 9, 675. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Canizares, C.; Jorrin, B.; Poole, P.S.; Tkacz, A. Understanding the holobiont: The interdependence of plants and their microbiome. Curr. Opin. Microbiol. 2017, 38, 188–196. [Google Scholar] [CrossRef]
- Rosenberg, E. Holistic Fitness: Microbiomes are Part of the Holobiont’s Fitness. In Microbiomes; Springer: Berlin/Heidelberg, Germany, 2021; pp. 101–160. [Google Scholar]
- Vishwakarma, K.; Kumar, N.; Shandilya, C.; Mohapatra, S.; Bhayana, S.; Varma, A. Revisiting Plant-Microbe Interactions and Microbial Consortia Application for Enhancing Sustainable Agriculture: A Review. Front. Microbiol. 2020, 11, 560406. [Google Scholar] [CrossRef]
- Phour, M.; Sehrawat, A.; Sindhu, S.S.; Glick, B.R. Interkingdom signaling in plant-rhizomicrobiome interactions for sustainable agriculture. Microbiol. Res. 2020, 241, 126589. [Google Scholar] [CrossRef]
- Matyssek, R.; Lüttge, U.; zu Castell, W. Evolution of Holobiont-Like Systems: From Individual to Composed Ecological and Global Units. In Progress in Botany; Springer: Berlin/Heidelberg, Germany, 2022. [Google Scholar]
- Harris, J.M.; Balint-Kurti, P.; Bede, J.C.; Day, B.; Gold, S.; Goss, E.M.; Grenville-Briggs, L.J.; Jones, K.M.; Wang, A.; Wang, Y.; et al. What are the Top 10 Unanswered Questions in Molecular Plant-Microbe Interactions? Mol. Plant. Microbe Interact. 2020, 33, 1354–1365. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Michalski, A.; Damoc, E.; Hauschild, J.P.; Lange, O.; Wieghaus, A.; Makarov, A.; Nagaraj, N.; Cox, J.; Mann, M.; Horning, S. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell Proteom. 2011, 10, M111 011015. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Pierroz, G.; Wipf, H.M.; Gao, C.; Taylor, J.W.; Lemaux, P.G.; Coleman-Derr, D. Holo-omics for deciphering plant-microbiome interactions. Microbiome 2021, 9, 69. [Google Scholar] [CrossRef]
- Shade, A. Diversity is the question, not the answer. ISME J. 2017, 11, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, R.H.; Ryberg, M.; Abarenkov, K.; Sjokvist, E.; Kristiansson, E. The ITS region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiol. Lett. 2009, 296, 97–101. [Google Scholar] [CrossRef]
- Bellemain, E.; Carlsen, T.; Brochmann, C.; Coissac, E.; Taberlet, P.; Kauserud, H. ITS as an environmental DNA barcode for fungi: An in silico approach reveals potential PCR biases. BMC Microbiol. 2010, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- Begerow, D.; Nilsson, H.; Unterseher, M.; Maier, W. Current state and perspectives of fungal DNA barcoding and rapid identification procedures. Appl. Microbiol. Biotechnol. 2010, 87, 99–108. [Google Scholar] [CrossRef]
- Lucking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 14. [Google Scholar] [CrossRef]
- Shade, A.; Stopnisek, N. Abundance-occupancy distributions to prioritize plant core microbiome membership. Curr. Opin. Microbiol. 2019, 49, 50–58. [Google Scholar] [CrossRef]
- Shade, A.; Jones, S.E.; Caporaso, J.G.; Handelsman, J.; Knight, R.; Fierer, N.; Gilbert, J.A. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 2014, 5, e01371-14. [Google Scholar] [CrossRef] [Green Version]
- Beilsmith, K.; Thoen, M.P.M.; Brachi, B.; Gloss, A.D.; Khan, M.H.; Bergelson, J. Genome-wide association studies on the phyllosphere microbiome: Embracing complexity in host-microbe interactions. Plant J. 2019, 97, 164–181. [Google Scholar] [CrossRef] [Green Version]
- Meyer, F.; Fritz, A.; Deng, Z.L.; Koslicki, D.; Lesker, T.R.; Gurevich, A.; Robertson, G.; Alser, M.; Antipov, D.; Beghini, F.; et al. Critical Assessment of Metagenome Interpretation: The second round of challenges. Nat. Methods 2022, 19, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Shakya, M.; Lo, C.C.; Chain, P.S.G. Advances and Challenges in Metatranscriptomic Analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef] [Green Version]
- Van Den Bossche, T.; Kunath, B.J.; Schallert, K.; Schape, S.S.; Abraham, P.E.; Armengaud, J.; Arntzen, M.O.; Bassignani, A.; Benndorf, D.; Fuchs, S.; et al. Critical Assessment of MetaProteome Investigation (CAMPI): A multi-laboratory comparison of established workflows. Nat. Commun. 2021, 12, 7305. [Google Scholar] [CrossRef]
- Jacoby, R.P.; Chen, L.; Schwier, M.; Koprivova, A.; Kopriva, S. Recent advances in the role of plant metabolites in shaping the root microbiome. F1000Research 2020, 9, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roume, H.; Muller, E.E.; Cordes, T.; Renaut, J.; Hiller, K.; Wilmes, P. A biomolecular isolation framework for eco-systems biology. ISME J. 2013, 7, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.; Conway, J.M.; Dangl, J.L.; Woyke, T. Elucidating Bacterial Gene Functions in the Plant Microbiome. Cell Host Microbe 2018, 24, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Lambais, M.R.; Barrera, S.E.; Santos, E.C.; Crowley, D.E.; Jumpponen, A. Phyllosphere Metaproteomes of Trees from the Brazilian Atlantic Forest Show High Levels of Functional Redundancy. Microb. Ecol. 2017, 73, 123–134. [Google Scholar] [CrossRef]
- Abram, F. Systems-based approaches to unravel multi-species microbial community functioning. Comput. Struct. Biotechnol. J. 2015, 13, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Vayssier-Taussat, M.; Albina, E.; Citti, C.; Cosson, J.F.; Jacques, M.A.; Lebrun, M.H.; Le Loir, Y.; Ogliastro, M.; Petit, M.A.; Roumagnac, P.; et al. Shifting the paradigm from pathogens to pathobiome: New concepts in the light of meta-omics. Front. Cell. Infect. Microbiol. 2014, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K.; Lindon, J.C. Systems biology: Metabonomics. Nature 2008, 455, 1054–1056. [Google Scholar] [CrossRef]
- Ghannam, R.B.; Techtmann, S.M. Machine learning applications in microbial ecology, human microbiome studies, and environmental monitoring. Comput. Struct. Biotechnol. J. 2021, 19, 1092–1107. [Google Scholar] [CrossRef]
- Leite, M.F.; Kuramae, E.E. You must choose, but choose wisely: Model-based approaches for microbial community analysis. Soil Biol. Biochem. 2020, 151, 108042. [Google Scholar] [CrossRef]
- Tecon, R.; Mitri, S.; Ciccarese, D.; Or, D.; van der Meer, J.R.; Johnson, D.R. Bridging the Holistic-Reductionist Divide in Microbial Ecology. mSystems 2019, 4, e00265-18. [Google Scholar] [CrossRef] [Green Version]
- Swenson, N.G.; Jones, F.A. Community transcriptomics, genomics and the problem of species co-occurrence. J. Ecol. 2017, 105, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Zengler, K.; Hofmockel, K.; Baliga, N.S.; Behie, S.W.; Bernstein, H.C.; Brown, J.B.; Dinneny, J.R.; Floge, S.A.; Forry, S.P.; Hess, M.; et al. EcoFABs: Advancing microbiome science through standardized fabricated ecosystems. Nat. Methods 2019, 16, 567–571. [Google Scholar] [CrossRef]
- Oyserman, B.O.; Cordovez, V.; Flores, S.S.; Leite, M.F.A.; Nijveen, H.; Medema, M.H.; Raaijmakers, J.M. Extracting the GEMs: Genotype, Environment, and Microbiome Interactions Shaping Host Phenotypes. Front. Microbiol. 2020, 11, 574053. [Google Scholar] [CrossRef]
- Stopnisek, N.; Shade, A. Persistent microbiome members in the common bean rhizosphere: An integrated analysis of space, time, and plant genotype. ISME J. 2021, 15, 2708–2722. [Google Scholar] [CrossRef]
- Raes, J.; Letunic, I.; Yamada, T.; Jensen, L.J.; Bork, P. Toward molecular trait-based ecology through integration of biogeochemical, geographical and metagenomic data. Mol. Syst. Biol. 2011, 7, 473. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, C.R.; Salas-Gonzalez, I.; Conway, J.M.; Finkel, O.M.; Gilbert, S.; Russ, D.; Teixeira, P.; Dangl, J.L. The Plant Microbiome: From Ecology to Reductionism and Beyond. Annu. Rev. Microbiol. 2020, 74, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Larrainzar, E.; Riely, B.K.; Kim, S.C.; Carrasquilla-Garcia, N.; Yu, H.J.; Hwang, H.J.; Oh, M.; Kim, G.B.; Surendrarao, A.K.; Chasman, D.; et al. Deep Sequencing of the Medicago truncatula Root Transcriptome Reveals a Massive and Early Interaction between Nodulation Factor and Ethylene Signals. Plant Physiol. 2015, 169, 233–265. [Google Scholar] [CrossRef] [Green Version]
- Petre, B.; Lorrain, C.; Stukenbrock, E.H.; Duplessis, S. Host-specialized transcriptome of plant-associated organisms. Curr. Opin. Plant Biol. 2020, 56, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Tabrett, A.; Horton, M.W. The influence of host genetics on the microbiome. F1000Research 2020, 9, 84. [Google Scholar] [CrossRef]
- Clouse, K.M.; Wagner, M.R. Plant Genetics as a Tool for Manipulating Crop Microbiomes: Opportunities and Challenges. Front. Bioeng. Biotechnol. 2021, 9, 567548. [Google Scholar] [CrossRef]
- Lebeis, S.L.; Paredes, S.H.; Lundberg, D.S.; Breakfield, N.; Gehring, J.; McDonald, M.; Malfatti, S.; Glavina del Rio, T.; Jones, C.D.; Tringe, S.G.; et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 2015, 349, 860–864. [Google Scholar] [CrossRef] [Green Version]
- Bodenhausen, N.; Bortfeld-Miller, M.; Ackermann, M.; Vorholt, J.A. A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genet. 2014, 10, e1004283. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Z.; Yates, T.B.; Shrestha, H.K.; Engle, N.L.; Flanagan, A.; Morrell-Falvey, J.L.; Sun, Y.; Tschaplinski, T.J.; Abraham, P.E.; Labbe, J.; et al. Towards engineering ectomycorrhization into switchgrass bioenergy crops via a lectin receptor-like kinase. Plant Biotechnol. J. 2021, 19, 2454–2468. [Google Scholar] [CrossRef]
- Edwards, J.A.; Santos-Medellin, C.M.; Liechty, Z.S.; Nguyen, B.; Lurie, E.; Eason, S.; Phillips, G.; Sundaresan, V. Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 2018, 16, e2003862. [Google Scholar] [CrossRef]
- Wagner, M.R.; Lundberg, D.S.; Del Rio, T.G.; Tringe, S.G.; Dangl, J.L.; Mitchell-Olds, T. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 2016, 7, 12151. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.T.; Wagner, M.R.; Rushworth, C.A.; Prasad, K.V.; Mitchell-Olds, T. The evolution of quantitative traits in complex environments. Heredity 2014, 112, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Horton, M.W.; Bodenhausen, N.; Beilsmith, K.; Meng, D.; Muegge, B.D.; Subramanian, S.; Vetter, M.M.; Vilhjalmsson, B.J.; Nordborg, M.; Gordon, J.I.; et al. Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nat. Commun. 2014, 5, 5320. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Nomura, K.; Wang, X.; Sohrabi, R.; Xu, J.; Yao, L.; Paasch, B.C.; Ma, L.; Kremer, J.; Cheng, Y.; et al. A plant genetic network for preventing dysbiosis in the phyllosphere. Nature 2020, 580, 653–657. [Google Scholar] [CrossRef]
- Xiong, C.; Zhu, Y.G.; Wang, J.T.; Singh, B.; Han, L.L.; Shen, J.P.; Li, P.P.; Wang, G.B.; Wu, C.F.; Ge, A.H.; et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2021, 229, 1091–1104. [Google Scholar] [CrossRef]
- Henry, L.P.; Bruijning, M.; Forsberg, S.K.G.; Ayroles, J.F. The microbiome extends host evolutionary potential. Nat. Commun. 2021, 12, 5141. [Google Scholar] [CrossRef] [PubMed]
- Laforest-Lapointe, I.; Paquette, A.; Messier, C.; Kembel, S.W. Leaf bacterial diversity mediates plant diversity and ecosystem function relationships. Nature 2017, 546, 145–147. [Google Scholar] [CrossRef]
- Diakite, A.; Dubourg, G.; Dione, N.; Afouda, P.; Bellali, S.; Ngom, I.I.; Valles, C.; Tall, M.L.; Lagier, J.C.; Raoult, D. Optimization and standardization of the culturomics technique for human microbiome exploration. Sci. Rep. 2020, 10, 9674. [Google Scholar] [CrossRef]
- Regalado, J.; Lundberg, D.S.; Deusch, O.; Kersten, S.; Karasov, T.; Poersch, K.; Shirsekar, G.; Weigel, D. Combining whole-genome shotgun sequencing and rRNA gene amplicon analyses to improve detection of microbe-microbe interaction networks in plant leaves. ISME J. 2020, 14, 2116–2130. [Google Scholar] [CrossRef]
- Niu, B.; Paulson, J.N.; Zheng, X.; Kolter, R. Simplified and representative bacterial community of maize roots. Proc. Natl. Acad. Sci. USA 2017, 114, E2450–E2459. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Wicaksono, W.A.; Cernava, T.; Berg, C.; Berg, G. Bog ecosystems as a playground for plant-microbe coevolution: Bryophytes and vascular plants harbour functionally adapted bacteria. Microbiome 2021, 9, 170. [Google Scholar] [CrossRef]
- Zheng, Y.; Xu, Z.; Liu, H.; Liu, Y.; Zhou, Y.; Meng, C.; Ma, S.; Xie, Z.; Li, Y.; Zhang, C.S. Patterns in the Microbial Community of Salt-Tolerant Plants and the Functional Genes Associated with Salt Stress Alleviation. Microbiol. Spectr. 2021, 9, e0076721. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Chiniquy, D.; Pierroz, G.; Deng, S.; Gao, C.; Diamond, S.; Simmons, T.; Wipf, H.M.; Caddell, D.; et al. Genome-resolved metagenomics reveals role of iron metabolism in drought-induced rhizosphere microbiome dynamics. Nat. Commun. 2021, 12, 3209. [Google Scholar] [CrossRef]
- Starr, E.P.; Shi, S.; Blazewicz, S.J.; Probst, A.J.; Herman, D.J.; Firestone, M.K.; Banfield, J.F. Stable isotope informed genome-resolved metagenomics reveals that Saccharibacteria utilize microbially-processed plant-derived carbon. Microbiome 2018, 6, 122. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Chai, X.; Zhang, L.; George, T.S.; Wang, F.; Feng, G. Different Arbuscular Mycorrhizal Fungi Cocolonizing on a Single Plant Root System Recruit Distinct Microbiomes. mSystems 2020, 5, e00929-20. [Google Scholar] [CrossRef]
- Starr, E.P.; Shi, S.; Blazewicz, S.J.; Koch, B.J.; Probst, A.J.; Hungate, B.A.; Pett-Ridge, J.; Firestone, M.K.; Banfield, J.F. Stable-Isotope-Informed, Genome-Resolved Metagenomics Uncovers Potential Cross-Kingdom Interactions in Rhizosphere Soil. mSphere 2021, 6, e0008521. [Google Scholar] [CrossRef]
- Mark, G.L.; Dow, J.M.; Kiely, P.D.; Higgins, H.; Haynes, J.; Baysse, C.; Abbas, A.; Foley, T.; Franks, A.; Morrissey, J.; et al. Transcriptome profiling of bacterial responses to root exudates identifies genes involved in microbe-plant interactions. Proc. Natl. Acad. Sci. USA 2005, 102, 17454–17459. [Google Scholar] [CrossRef] [Green Version]
- Spaepen, S.; Bossuyt, S.; Engelen, K.; Marchal, K.; Vanderleyden, J. Phenotypical and molecular responses of Arabidopsis thaliana roots as a result of inoculation with the auxin-producing bacterium Azospirillum brasilense. New Phytol. 2014, 201, 850–861. [Google Scholar] [CrossRef]
- Santos-Medellin, C.; Edwards, J.; Nguyen, B.; Sundaresan, V. Acquisition of a complex root microbiome reshapes the transcriptomes of rice plants. New Phytol. 2022, 235, 2008–2021. [Google Scholar] [CrossRef]
- Nerva, L.; Garcia, J.F.; Favaretto, F.; Giudice, G.; Moffa, L.; Sandrini, M.; Cantu, D.; Zanzotto, A.; Gardiman, M.; Velasco, R.; et al. The hidden world within plants: Metatranscriptomics unveils the complexity of wood microbiomes. J. Exp. Bot. 2022, 73, 2682–2697. [Google Scholar] [CrossRef]
- Yamazaki, S.; Mardani-Korrani, H.; Kaida, R.; Ochiai, K.; Kobayashi, M.; Nagano, A.J.; Fujii, Y.; Sugiyama, A.; Aoki, Y. Field multi-omics analysis reveals a close association between bacterial communities and mineral properties in the soybean rhizosphere. Sci. Rep. 2021, 11, 8878. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Wang, E.; Wang, N. Mechanisms Underlying the Rhizosphere-To-Rhizoplane Enrichment of Cellvibrio Unveiled by Genome-Centric Metagenomics and Metatranscriptomics. Microorganisms 2020, 8, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, E.; Pitre, F.E.; Page, A.P.; Marleau, J.; Guidi Nissim, W.; St-Arnaud, M.; Labrecque, M.; Joly, S.; Yergeau, E.; Brereton, N.J.B. Trees, fungi and bacteria: Tripartite metatranscriptomics of a root microbiome responding to soil contamination. Microbiome 2018, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, S.; Kaneko, T.; Okubo, T.; Rallos, L.E.; Eda, S.; Mitsui, H.; Sato, S.; Nakamura, Y.; Tabata, S.; Minamisawa, K. Development of a bacterial cell enrichment method and its application to the community analysis in soybean stems. Microb. Ecol. 2009, 58, 703–714. [Google Scholar] [CrossRef]
- Jiao, J.Y.; Wang, H.X.; Zeng, Y.; Shen, Y.M. Enrichment for microbes living in association with plant tissues. J. Appl. Microbiol. 2006, 100, 830–837. [Google Scholar] [CrossRef]
- Song, L.; Xie, K. Engineering CRISPR/Cas9 to mitigate abundant host contamination for 16S rRNA gene-based amplicon sequencing. Microbiome 2020, 8, 80. [Google Scholar] [CrossRef]
- Nobori, T.; Tsuda, K. In planta Transcriptome Analysis of Pseudomonas syringae. Bio Protoc. 2018, 8, e2987. [Google Scholar] [CrossRef]
- Garcia, B.J.; Simha, R.; Garvin, M.; Furches, A.; Jones, P.; Gazolla, J.; Hyatt, P.D.; Schadt, C.W.; Pelletier, D.; Jacobson, D. A k-mer based approach for classifying viruses without taxonomy identifies viral associations in human autism and plant microbiomes. Comput. Struct. Biotechnol. J. 2021, 19, 5911–5919. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, A.; Leclercq, M.; Sanabria, M.; Perin, O.; Droit, A. Machine Learning and Deep Learning Applications in Metagenomic Taxonomy and Functional Annotation. Front. Microbiol. 2022, 13, 811495. [Google Scholar] [CrossRef]
- Ghurye, J.S.; Cepeda-Espinoza, V.; Pop, M. Metagenomic Assembly: Overview, Challenges and Applications. Yale J. Biol. Med. 2016, 89, 353–362. [Google Scholar]
- Nissen, J.N.; Johansen, J.; Allesoe, R.L.; Sonderby, C.K.; Armenteros, J.J.A.; Gronbech, C.H.; Jensen, L.J.; Nielsen, H.B.; Petersen, T.N.; Winther, O.; et al. Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 2021, 39, 555–560. [Google Scholar] [CrossRef]
- Alberdi, A.; Andersen, S.B.; Limborg, M.T.; Dunn, R.R.; Gilbert, M.T.P. Disentangling host-microbiota complexity through hologenomics. Nat. Rev. Genet. 2022, 23, 281–297. [Google Scholar] [CrossRef]
- Feussner, I.; Polle, A. What the transcriptome does not tell—Proteomics and metabolomics are closer to the plants’ patho-phenotype. Curr. Opin. Plant Biol. 2015, 26, 26–31. [Google Scholar] [CrossRef]
- Smith, L.M.; Kelleher, N.L. Proteoforms as the next proteomics currency. Science 2018, 359, 1106–1107. [Google Scholar] [CrossRef]
- Smith, L.M.; Agar, J.N.; Chamot-Rooke, J.; Danis, P.O.; Ge, Y.; Loo, J.A.; Pasa-Tolic, L.; Tsybin, Y.O.; Kelleher, N.L.; Consortium for Top-Down, P. The Human Proteoform Project: Defining the human proteome. Sci. Adv. 2021, 7, eabk0734. [Google Scholar] [CrossRef]
- Mergner, J.; Frejno, M.; List, M.; Papacek, M.; Chen, X.; Chaudhary, A.; Samaras, P.; Richter, S.; Shikata, H.; Messerer, M.; et al. Mass-spectrometry-based draft of the Arabidopsis proteome. Nature 2020, 579, 409–414. [Google Scholar] [CrossRef]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Marx, H.; Minogue, C.E.; Jayaraman, D.; Richards, A.L.; Kwiecien, N.W.; Siahpirani, A.F.; Rajasekar, S.; Maeda, J.; Garcia, K.; Del Valle-Echevarria, A.R.; et al. A proteomic atlas of the legume Medicago truncatula and its nitrogen-fixing endosymbiont Sinorhizobium meliloti. Nat. Biotechnol. 2016, 34, 1198–1205. [Google Scholar] [CrossRef]
- Sebastiana, M.; Martins, J.; Figueiredo, A.; Monteiro, F.; Sardans, J.; Penuelas, J.; Silva, A.; Roepstorff, P.; Pais, M.S.; Coelho, A.V. Oak protein profile alterations upon root colonization by an ectomycorrhizal fungus. Mycorrhiza 2017, 27, 109–128. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, D.; Forshey, K.L.; Grimsrud, P.A.; Ane, J.M. Leveraging proteomics to understand plant-microbe interactions. Front. Plant Sci. 2012, 3, 44. [Google Scholar] [CrossRef]
- Khatabi, B.; Gharechahi, J.; Ghaffari, M.R.; Liu, D.; Haynes, P.A.; McKay, M.J.; Mirzaei, M.; Salekdeh, G.H. Plant-Microbe Symbiosis: What Has Proteomics Taught Us? Proteomics 2019, 19, e1800105. [Google Scholar] [CrossRef]
- Elmore, J.M.; Liu, J.; Smith, B.; Phinney, B.; Coaker, G. Quantitative proteomics reveals dynamic changes in the plasma membrane during Arabidopsis immune signaling. Mol. Cell. Proteom. 2012, 11, M111.014555. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.M.; Thomas, V.; Bennett, M.H.; Mansfield, J.; Grant, M. Modifications to the Arabidopsis defense proteome occur prior to significant transcriptional change in response to inoculation with Pseudomonas syringae. Plant Physiol. 2006, 142, 1603–1620. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Saravanan, R.S.; Damasceno, C.M.; Yamane, H.; Kim, B.D.; Rose, J.K. Digging deeper into the plant cell wall proteome. Plant Physiol. Biochem. 2004, 42, 979–988. [Google Scholar] [CrossRef]
- Shrivastava, N.; Jiang, L.; Li, P.; Sharma, A.K.; Luo, X.; Wu, S.; Pandey, R.; Gao, Q.; Lou, B. Proteomic approach to understand the molecular physiology of symbiotic interaction between Piriformospora indica and Brassica napus. Sci. Rep. 2018, 8, 5773. [Google Scholar] [CrossRef]
- Rose, C.M.; Venkateshwaran, M.; Volkening, J.D.; Grimsrud, P.A.; Maeda, J.; Bailey, D.J.; Park, K.; Howes-Podoll, M.; den Os, D.; Yeun, L.H.; et al. Rapid phosphoproteomic and transcriptomic changes in the rhizobia-legume symbiosis. Mol. Cell. Proteom. 2012, 11, 724–744. [Google Scholar] [CrossRef] [Green Version]
- Riley, N.M.; Coon, J.J. Phosphoproteomics in the Age of Rapid and Deep Proteome Profiling. Anal. Chem. 2016, 88, 74–94. [Google Scholar] [CrossRef]
- Benschop, J.J.; Mohammed, S.; O’Flaherty, M.; Heck, A.J.; Slijper, M.; Menke, F.L. Quantitative phosphoproteomics of early elicitor signaling in Arabidopsis. Mol. Cell. Proteom. 2007, 6, 1198–1214. [Google Scholar] [CrossRef] [Green Version]
- Rayapuram, N.; Bigeard, J.; Alhoraibi, H.; Bonhomme, L.; Hesse, A.M.; Vinh, J.; Hirt, H.; Pflieger, D. Quantitative Phosphoproteomic Analysis Reveals Shared and Specific Targets of Arabidopsis Mitogen-Activated Protein Kinases (MAPKs) MPK3, MPK4, and MPK6. Mol. Cell. Proteom. 2018, 17, 61–80. [Google Scholar] [CrossRef] [Green Version]
- Nakagami, H.; Sugiyama, N.; Mochida, K.; Daudi, A.; Yoshida, Y.; Toyoda, T.; Tomita, M.; Ishihama, Y.; Shirasu, K. Large-scale comparative phosphoproteomics identifies conserved phosphorylation sites in plants. Plant Physiol. 2010, 153, 1161–1174. [Google Scholar] [CrossRef]
- Long, S.; Yang, Y.; Shen, C.; Wang, Y.; Deng, A.; Qin, Q.; Qiao, L. Metaproteomics characterizes human gut microbiome function in colorectal cancer. NPJ Biofilms Microbiomes 2020, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, W.; Abraham, P.E.; Li, Z.; Pan, C.; Hettich, R.L. Microbial metaproteomics for characterizing the range of metabolic functions and activities of human gut microbiota. Proteomics 2015, 15, 3424–3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmes, P.; Heintz-Buschart, A.; Bond, P.L. A decade of metaproteomics: Where we stand and what the future holds. Proteomics 2015, 15, 3409–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Z.; Okubo, T.; Kubota, K.; Kasahara, Y.; Tsurumaru, H.; Anda, M.; Ikeda, S.; Minamisawa, K. Metaproteomic identification of diazotrophic methanotrophs and their localization in root tissues of field-grown rice plants. Appl. Environ. Microbiol. 2014, 80, 5043–5052. [Google Scholar] [CrossRef] [Green Version]
- Knief, C.; Delmotte, N.; Chaffron, S.; Stark, M.; Innerebner, G.; Wassmann, R.; von Mering, C.; Vorholt, J.A. Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J. 2012, 6, 1378–1390. [Google Scholar] [CrossRef] [Green Version]
- Salvato, F.; Vintila, S.; Finkel, O.M.; Dangl, J.; Kleiner, M. Evaluation of protein extraction methods for metaproteomic analyses of root-associated microbes. Mol. Plant Microbe Interact. 2022. [Google Scholar] [CrossRef]
- Appidi, M.R.; Bible, A.N.; Carper, D.L.; Jawdy, S.S.; Giannone, R.J.; Hettich, R.L.; Morrell-Falvey, J.; Abraham, P.E. Development of an Experimental Approach to Achieve Spatially Resolved Plant Root-Associated Metaproteomics Using an Agar-Plate System. Mol. Plant Microbe Interact. 2022, 35, 639–649. [Google Scholar] [CrossRef]
- Shrestha, H.K.; Appidi, M.R.; Villalobos Solis, M.I.; Wang, J.; Carper, D.L.; Burdick, L.; Pelletier, D.A.; Doktycz, M.J.; Hettich, R.L.; Abraham, P.E. Metaproteomics reveals insights into microbial structure, interactions, and dynamic regulation in defined communities as they respond to environmental disturbance. BMC Microbiol. 2021, 21, 308. [Google Scholar] [CrossRef]
- Chourey, K.; Jansson, J.; VerBerkmoes, N.; Shah, M.; Chavarria, K.L.; Tom, L.M.; Brodie, E.L.; Hettich, R.L. Direct cellular lysis/protein extraction protocol for soil metaproteomics. J. Proteome Res. 2010, 9, 6615–6622. [Google Scholar] [CrossRef]
- Qian, C.; Hettich, R.L. Optimized Extraction Method to Remove Humic Acid Interferences from Soil Samples Prior to Microbial Proteome Measurements. J. Proteome Res. 2017, 16, 2537–2546. [Google Scholar] [CrossRef] [PubMed]
- Mandalakis, M.; Panikov, N.S.; Polymenakou, P.N.; Sizova, M.V.; Stamatakis, A. A simple cleanup method for the removal of humic substances from soil protein extracts using aluminum coagulation. Environ. Sci. Pollut. Res. Int. 2018, 25, 23845–23856. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Bastida, F.; Sciarrillo, R.; Guarino, C. Soil Metaproteomics for the Study of the Relationships Between Microorganisms and Plants: A Review of Extraction Protocols and Ecological Insights. Int. J. Mol. Sci. 2020, 21, 8455. [Google Scholar] [CrossRef]
- Kleiner, M.; Thorson, E.; Sharp, C.E.; Dong, X.; Liu, D.; Li, C.; Strous, M. Assessing species biomass contributions in microbial communities via metaproteomics. Nat. Commun. 2017, 8, 1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouffret, V.; Miotello, G.; Culotta, K.; Ayrault, S.; Pible, O.; Armengaud, J. Increasing the power of interpretation for soil metaproteomics data. Microbiome 2021, 9, 195. [Google Scholar] [CrossRef] [PubMed]
- Bauermeister, A.; Mannochio-Russo, H.; Costa-Lotufo, L.V.; Jarmusch, A.K.; Dorrestein, P.C. Mass spectrometry-based metabolomics in microbiome investigations. Nat. Rev. Microbiol. 2022, 20, 143–160. [Google Scholar] [CrossRef]
- Aksenov, A.A.; da Silva, R.; Knight, R.; Lopes, N.P.; Dorrestein, P.C. Global chemical analysis of biology by mass spectrometry. Nat. Rev. Chem. 2017, 1, 0054. [Google Scholar] [CrossRef]
- Sasse, J.; Martinoia, E.; Northen, T. Feed Your Friends: Do Plant Exudates Shape the Root Microbiome? Trends Plant Sci. 2018, 23, 25–41. [Google Scholar] [CrossRef] [Green Version]
- Hiruma, K. Roles of Plant-Derived Secondary Metabolites during Interactions with Pathogenic and Beneficial Microbes under Conditions of Environmental Stress. Microorganisms 2019, 7, 362. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Schillaci, M.; Roessner, U. Metabolomics as an emerging tool to study plant-microbe interactions. Emerg. Top. Life Sci. 2022, 6, 175–183. [Google Scholar] [CrossRef]
- Oburger, E.; Jones, D.L. Sampling root exudates–mission impossible? Rhizosphere 2018, 6, 116–133. [Google Scholar] [CrossRef]
- Kawasaki, A.; Okada, S.; Zhang, C.; Delhaize, E.; Mathesius, U.; Richardson, A.E.; Watt, M.; Gilliham, M.; Ryan, P.R. A sterile hydroponic system for characterising root exudates from specific root types and whole-root systems of large crop plants. Plant Methods 2018, 14, 114. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Guerrero, M.G.; Wang, P.; Phares, F.; Schachtman, D.P.; Alvarez, S.; van Dijk, K. A glass bead semi-hydroponic system for intact maize root exudate analysis and phenotyping. Plant Methods 2022, 18, 25. [Google Scholar] [CrossRef]
- Sasse, J.; Kant, J.; Cole, B.J.; Klein, A.P.; Arsova, B.; Schlaepfer, P.; Gao, J.; Lewald, K.; Zhalnina, K.; Kosina, S.; et al. Multilab EcoFAB study shows highly reproducible physiology and depletion of soil metabolites by a model grass. New Phytol. 2019, 222, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, J.; Schmidt, S.; Chutia, R.; Muller, J.; Bottcher, C.; Strehmel, N.; Scheel, D.; Abel, S. Non-targeted profiling of semi-polar metabolites in Arabidopsis root exudates uncovers a role for coumarin secretion and lignification during the local response to phosphate limitation. J. Exp. Bot. 2016, 67, 1421–1432. [Google Scholar] [CrossRef]
- Chaparro, J.M.; Badri, D.V.; Bakker, M.G.; Sugiyama, A.; Manter, D.K.; Vivanco, J.M. Root exudation of phytochemicals in Arabidopsis follows specific patterns that are developmentally programmed and correlate with soil microbial functions. PLoS ONE 2013, 8, e55731. [Google Scholar] [CrossRef]
- Strehmel, N.; Bottcher, C.; Schmidt, S.; Scheel, D. Profiling of secondary metabolites in root exudates of Arabidopsis thaliana. Phytochemistry 2014, 108, 35–46. [Google Scholar] [CrossRef]
- Carvalhais, L.C.; Dennis, P.G.; Fedoseyenko, D.; Hajirezaei, M.R.; Borriss, R.; von Wirén, N. Root exudation of sugars, amino acids, and organic acids by maize as affected by nitrogen, phosphorus, potassium, and iron deficiency. J. Plant Nutr. Soil Sci. 2011, 174, 3–11. [Google Scholar] [CrossRef]
- Suzuki, K.; Okazaki, K.; Tawaraya, K.; Osaki, M.; Shinano, T. Gas chromatography–mass spectrometry associated global analysis of rice root exudates under aseptical conditions. Soil Sci. Plant Nutr. 2009, 55, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Tawaraya, K.; Horie, R.; Wagatsuma, T.; Saito, K.; Oikawa, A. Metabolite profiling of shoot extract, root extract, and root exudate of rice under nitrogen and phosphorus deficiency. Soil Sci. Plant Nutr. 2018, 64, 312–322. [Google Scholar] [CrossRef]
- Stringlis, I.A.; Yu, K.; Feussner, K.; de Jonge, R.; Van Bentum, S.; Van Verk, M.C.; Berendsen, R.L.; Bakker, P.; Feussner, I.; Pieterse, C.M.J. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl. Acad. Sci. USA 2018, 115, E5213–E5222. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, D.; Wang, D.; Miao, Y.; Shao, J.; Zhou, X.; Xu, Z.; Li, Q.; Feng, H.; Li, S.; et al. Whole transcriptomic analysis of the plant-beneficial rhizobacterium Bacillus amyloliquefaciens SQR9 during enhanced biofilm formation regulated by maize root exudates. BMC Genom. 2015, 16, 685. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Robert, C.A.M.; Cadot, S.; Zhang, X.; Ye, M.; Li, B.; Manzo, D.; Chervet, N.; Steinger, T.; van der Heijden, M.G.A.; et al. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 2018, 9, 2738. [Google Scholar] [CrossRef] [Green Version]
- Phillips, R.P.; Erlitz, Y.; Bier, R.; Bernhardt, E.S. New approach for capturing soluble root exudates in forest soils. Funct. Ecol. 2008, 22, 990–999. [Google Scholar] [CrossRef]
- Wilson, R.M.; Tfaily, M.M.; Kolton, M.; Johnston, E.R.; Petro, C.; Zalman, C.A.; Hanson, P.J.; Heyman, H.M.; Kyle, J.E.; Hoyt, D.W.; et al. Soil metabolome response to whole-ecosystem warming at the Spruce and Peatland Responses under Changing Environments experiment. Proc. Natl. Acad. Sci. USA 2021, 118, e2004192118. [Google Scholar] [CrossRef]
- Li, Y.; Xu, L.; Letuma, P.; Lin, W. Metabolite profiling of rhizosphere soil of different allelopathic potential rice accessions. BMC Plant Biol. 2020, 20, 265. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, Y.; Ling, W.; Kong, H.; Zhu, X. Gradient distribution of root exudates and polycyclic aromatic hydrocarbons in rhizosphere soil. Soil Sci. Soc. Am. J. 2011, 75, 1694–1703. [Google Scholar] [CrossRef]
- Petriacq, P.; Williams, A.; Cotton, A.; McFarlane, A.E.; Rolfe, S.A.; Ton, J. Metabolite profiling of non-sterile rhizosphere soil. Plant J. 2017, 92, 147–162. [Google Scholar] [CrossRef] [Green Version]
- Harman, G.; Khadka, R.; Doni, F.; Uphoff, N. Benefits to Plant Health and Productivity from Enhancing Plant Microbial Symbionts. Front. Plant Sci. 2020, 11, 610065. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Vivanco, J.M.; Shen, Q. The unseen rhizosphere root-soil-microbe interactions for crop production. Curr. Opin. Microbiol. 2017, 37, 8–14. [Google Scholar] [CrossRef]
- Huang, W.; Sun, D.; Chen, L.; An, Y. Integrative analysis of the microbiome and metabolome in understanding the causes of sugarcane bitterness. Sci. Rep. 2021, 11, 6024. [Google Scholar] [CrossRef]
- Gust, A.A.; Biswas, R.; Lenz, H.D.; Rauhut, T.; Ranf, S.; Kemmerling, B.; Gotz, F.; Glawischnig, E.; Lee, J.; Felix, G.; et al. Bacteria-derived peptidoglycans constitute pathogen-associated molecular patterns triggering innate immunity in Arabidopsis. J. Biol. Chem. 2007, 282, 32338–32348. [Google Scholar] [CrossRef] [Green Version]
- Camanes, G.; Scalschi, L.; Vicedo, B.; Gonzalez-Bosch, C.; Garcia-Agustin, P. An untargeted global metabolomic analysis reveals the biochemical changes underlying basal resistance and priming in Solanum lycopersicum, and identifies 1-methyltryptophan as a metabolite involved in plant responses to Botrytis cinerea and Pseudomonas syringae. Plant J. 2015, 84, 125–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Tan, D.X.; Reiter, R.J.; Shi, H. Comparative metabolomic analysis highlights the involvement of sugars and glycerol in melatonin-mediated innate immunity against bacterial pathogen in Arabidopsis. Sci. Rep. 2015, 5, 15815. [Google Scholar] [CrossRef] [Green Version]
- Korenblum, E.; Dong, Y.; Szymanski, J.; Panda, S.; Jozwiak, A.; Massalha, H.; Meir, S.; Rogachev, I.; Aharoni, A. Rhizosphere microbiome mediates systemic root metabolite exudation by root-to-root signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 3874–3883. [Google Scholar] [CrossRef]
- Huberty, M.; Martis, B.; van Kampen, J.; Choi, Y.H.; Vrieling, K.; Klinkhamer, P.G.L.; Bezemer, T.M. Soil Inoculation Alters Leaf Metabolic Profiles in Genetically Identical Plants. J. Chem. Ecol. 2020, 46, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Pang, Q.; Zhang, T.; Wang, Y.; Kong, W.; Guan, Q.; Yan, X.; Chen, S. Metabolomics of Early Stage Plant Cell-Microbe Interaction Using Stable Isotope Labeling. Front. Plant Sci. 2018, 9, 760. [Google Scholar] [CrossRef] [Green Version]
- Chokkathukalam, A.; Kim, D.H.; Barrett, M.P.; Breitling, R.; Creek, D.J. Stable isotope-labeling studies in metabolomics: New insights into structure and dynamics of metabolic networks. Bioanalysis 2014, 6, 511–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chialva, M.; Salvioli di Fossalunga, A.; Daghino, S.; Ghignone, S.; Bagnaresi, P.; Chiapello, M.; Novero, M.; Spadaro, D.; Perotto, S.; Bonfante, P. Native soils with their microbiotas elicit a state of alert in tomato plants. New Phytol. 2018, 220, 1296–1308. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Lin, R.; Tan, M.; Yan, J.; Yan, S. The mycorrhizal-induced growth promotion and insect resistance reduction in Populus alba x P. berolinensis seedlings: A multi-omics study. Tree Physiol. 2022, 42, 1059–1069. [Google Scholar] [CrossRef]
- Carrell, A.A.; Velickovic, D.; Lawrence, T.J.; Bowen, B.P.; Louie, K.B.; Carper, D.L.; Chu, R.K.; Mitchell, H.D.; Orr, G.; Markillie, L.M.; et al. Novel metabolic interactions and environmental conditions mediate the boreal peatmoss-cyanobacteria mutualism. ISME J. 2022, 16, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sidharth, S.; Zeng, S.; Jiang, Y.; Chan, Y.O.; Lyu, Z.; McCubbin, T.; Mertz, R.; Sharp, R.E.; Joshi, T. Bioinformatics for plant and agricultural discoveries in the age of multiomics: A review and case study of maize nodal root growth under water deficit. Physiol. Plant 2022, 174, e13672. [Google Scholar] [CrossRef] [PubMed]
- Weighill, D.; Tschaplinski, T.J.; Tuskan, G.A.; Jacobson, D. Data Integration in Poplar: ’Omics Layers and Integration Strategies. Front. Genet. 2019, 10, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weighill, D.; Jones, P.; Bleker, C.; Ranjan, P.; Shah, M.; Zhao, N.; Martin, M.; DiFazio, S.; Macaya-Sanz, D.; Schmutz, J.; et al. Multi-Phenotype Association Decomposition: Unraveling Complex Gene-Phenotype Relationships. Front. Genet. 2019, 10, 417. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, A.; Mathe, E.; Merling, M.; Ma, Q.; Liu, B. Network analyses in microbiome based on high-throughput multi-omics data. Brief. Bioinform. 2021, 22, 1639–1655. [Google Scholar] [CrossRef]
- Jones, P.; Weighill, D.; Shah, M.; Climer, S.; Schmutz, J.; Sreedasyam, A.; Tuskan, G.; Jacobson, D. Network Modeling of Complex Data Sets. Methods Mol. Biol. 2020, 2096, 197–215. [Google Scholar] [CrossRef]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.N.; Petipas, R.H.; Cheeke, T.E.; Rowland, J.L.; Friesen, M.L. Microbial Inoculants: Silver Bullet or Microbial Jurassic Park? Trends Microbiol. 2021, 29, 299–308. [Google Scholar] [CrossRef]
- Moore, J.A.M.; Abraham, P.E.; Michener, J.K.; Muchero, W.; Cregger, M.A. Ecosystem consequences of introducing plant growth promoting rhizobacteria to managed systems and potential legacy effects. New Phytol. 2022, 234, 1914–1918. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carper, D.L.; Appidi, M.R.; Mudbhari, S.; Shrestha, H.K.; Hettich, R.L.; Abraham, P.E. The Promises, Challenges, and Opportunities of Omics for Studying the Plant Holobiont. Microorganisms 2022, 10, 2013. https://doi.org/10.3390/microorganisms10102013
Carper DL, Appidi MR, Mudbhari S, Shrestha HK, Hettich RL, Abraham PE. The Promises, Challenges, and Opportunities of Omics for Studying the Plant Holobiont. Microorganisms. 2022; 10(10):2013. https://doi.org/10.3390/microorganisms10102013
Chicago/Turabian StyleCarper, Dana L., Manasa R. Appidi, Sameer Mudbhari, Him K. Shrestha, Robert L. Hettich, and Paul E. Abraham. 2022. "The Promises, Challenges, and Opportunities of Omics for Studying the Plant Holobiont" Microorganisms 10, no. 10: 2013. https://doi.org/10.3390/microorganisms10102013