



SARS-CoV-2 Variants by Whole-Genome Sequencing in a University Hospital in Bangkok: First to Third COVID-19 Waves
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Sample Selection and Viral RNA Extraction
2.3. Collection of Demographic and Clinical Data
2.4. WGS and Phylogenetic Tree
2.4.1. WGS of the First Wave
2.4.2. WGS of the Second and Third Waves
2.4.3. Phylogenetic Tree
2.5. SARS-CoV-2 Lineage Classification
3. Result
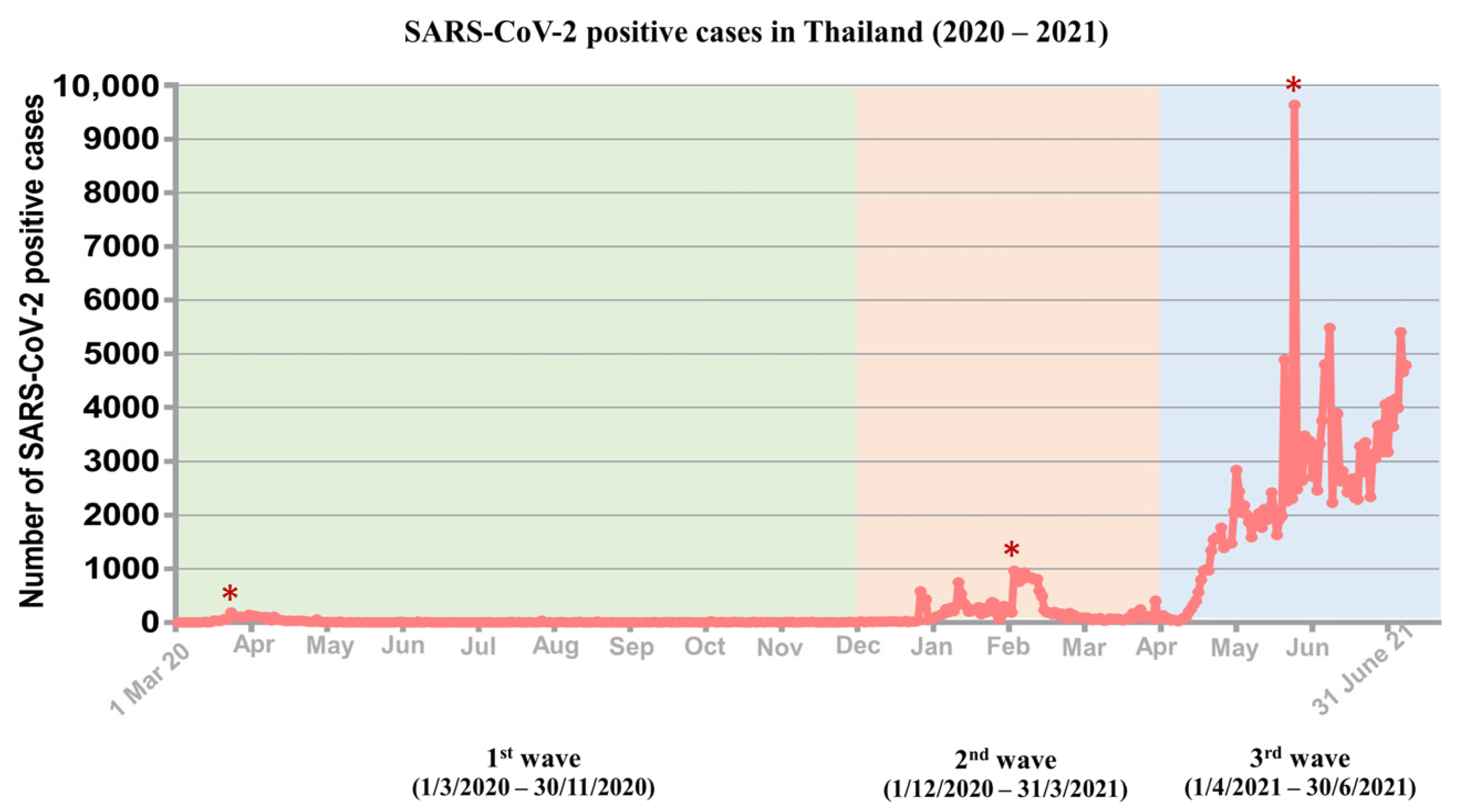
3.1. Distribution of Three Waves of COVID-19 Outbreaks in Thailand
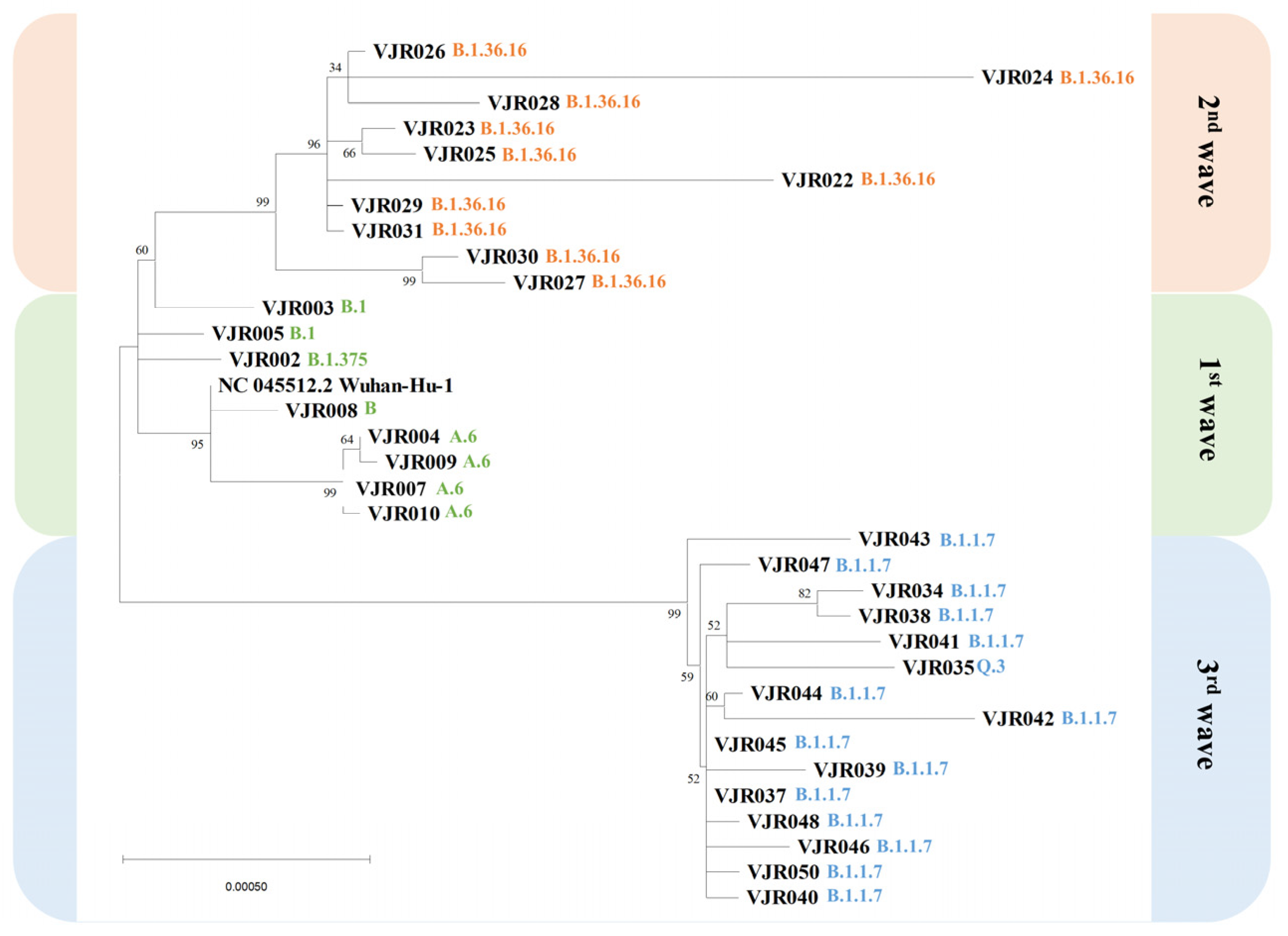
3.2. Phylogenetic Tree
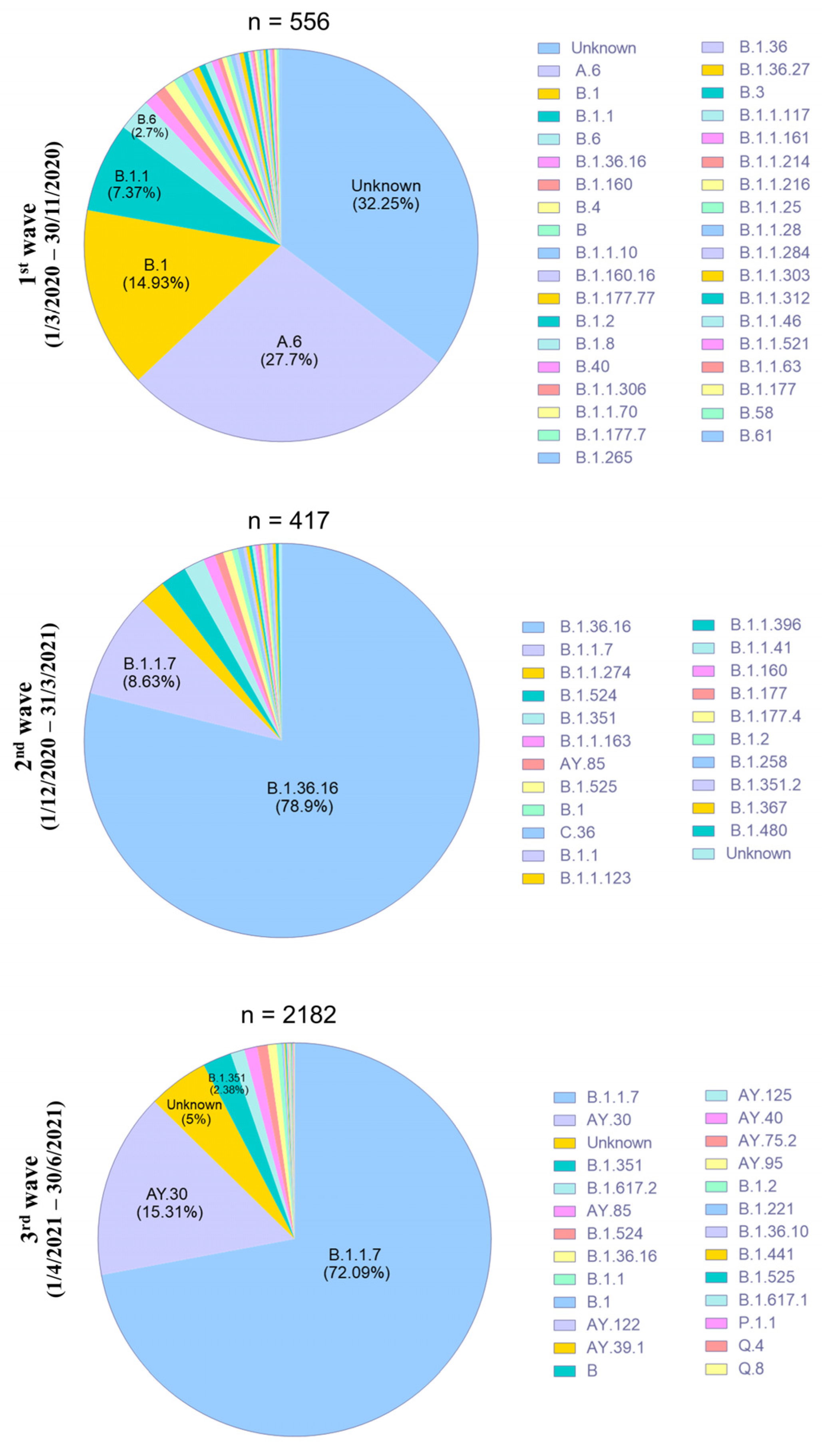
3.3. Characterization of SARS-CoV-2 Variants in Thailand
3.4. Clinical Characteristics of COVID-19 Patients in Three Epidemic Waves
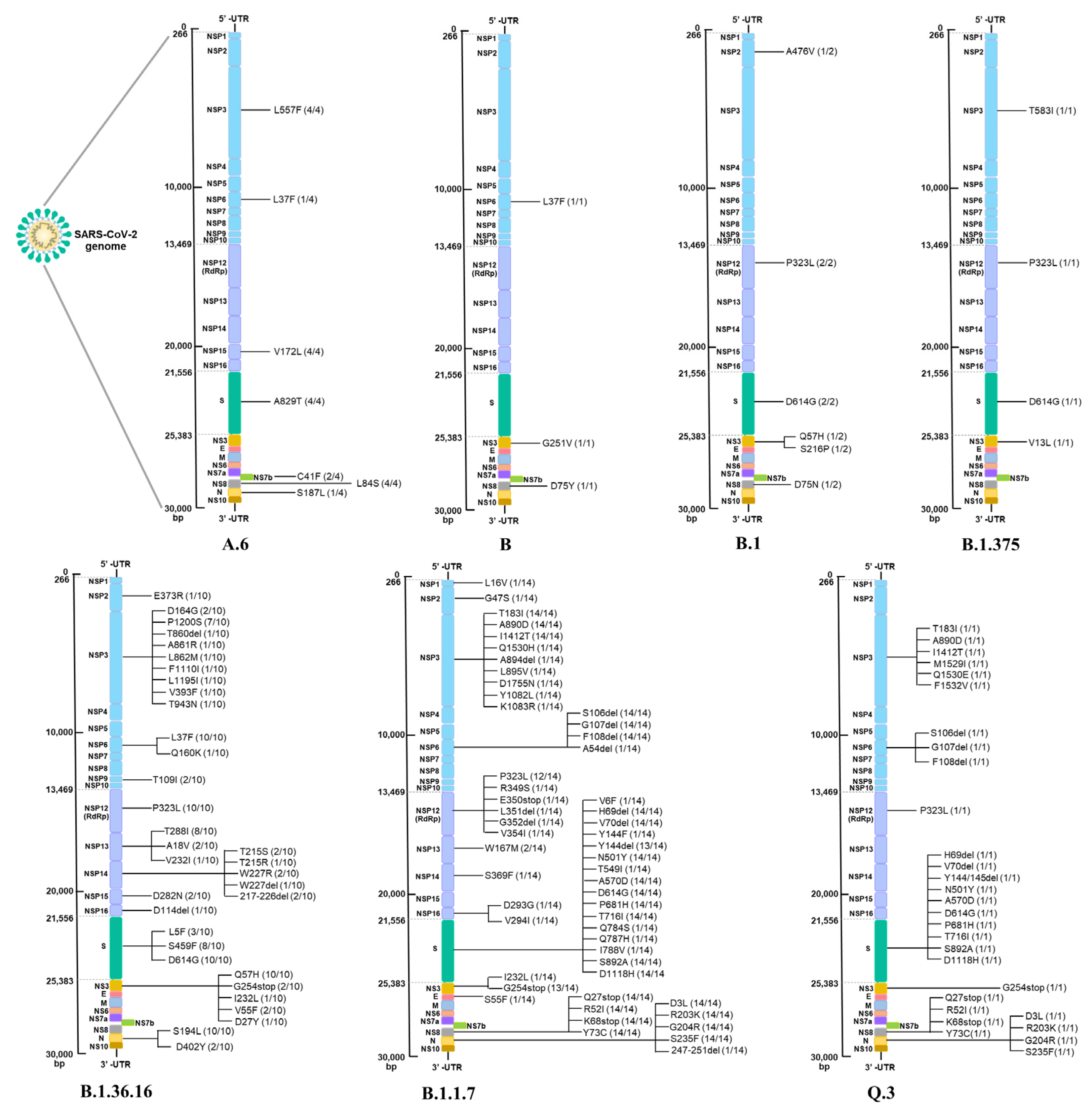
3.5. SARS-CoV-2 Mutations Associated with Disease Severity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 1st | first |
| 2nd | second |
| 3rd | third |
| aa | amino acid |
| ACE2 | angiotensin-converting enzyme 2 |
| cDNA | complementary DNA |
| COVID-19 | coronavirus disease 2019 |
| del | deletion |
| DNA | Deoxyribonucleic acid |
| GISAID | Global Initiative on Sharing Avian Influenza Data |
| NPS | nasopharyngeal swab |
| Nsp | nonstructural protein |
| RBD | receptor-binding domain |
| RNA | Ribonucleic acid |
| S protein | spike protein |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| ssCirDNA | circularized single strand DNA |
| VOC | variants of concern |
| VOI | variants of interest |
| WGS | whole-genome sequencing |
References
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 6 February 2023).
- Ghosh, N.; Saha, I.; Nandi, S.; Sharma, N. Characterisation of SARS-CoV-2 clades based on signature SNPs unveils continuous evolution. Methods 2021, 203, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Osman, I.O.; Levasseur, A.; Brechard, L.; Abdillahi Hassan, I.; Salah Abdillahi, I.; Ali Waberi, Z.; Delerce, J.; Bedotto, M.; Houhamdi, L.; Fournier, P.E.; et al. Whole Genome Sequencing of SARS-CoV-2 Strains in COVID-19 Patients from Djibouti Shows Novel Mutations and Clades Replacing over Time. Front. Med. 2021, 8, 737602. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Worp, N.; Nieuwenhuijse, D.F.; Sikkema, R.S.; Haagmans, B.; Fouchier, R.A.M.; Koopmans, M. The next phase of SARS-CoV-2 surveillance: Real-time molecular epidemiology. Nat. Med. 2021, 27, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.; Stowe, J.; Kirsebom, F.; Toffa, S.; Rickeard, T.; Gallagher, E.; Gower, C.; Kall, M.; Groves, N.; O’Connell, A.M.; et al. COVID-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 2022, 386, 1532–1546. [Google Scholar] [CrossRef] [PubMed]
- Buchan, S.A.; Chung, H.; Brown, K.A.; Austin, P.C.; Fell, D.B.; Gubbay, J.B.; Nasreen, S.; Schwartz, K.L.; Sundaram, M.E.; Tadrous, M.; et al. Estimated Effectiveness of COVID-19 Vaccines against Omicron or Delta Symptomatic Infection and Severe Outcomes. JAMA Netw. Open 2022, 5, e2232760. [Google Scholar] [CrossRef]
- Covariants. Shared Mutations Enabled by Data from GISAID. Available online: https://covariants.org/shared-mutations (accessed on 9 February 2023).
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D.; et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct. Target. Ther. 2022, 7, 138. [Google Scholar] [CrossRef]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard—Thailand Situation. Available online: https://covid19.who.int/region/searo/country/th (accessed on 6 February 2023).
- Kunno, J.; Supawattanabodee, B.; Sumanasrethakul, C.; Wiriyasivaj, B.; Kuratong, S.; Kaewchandee, C. Comparison of Different Waves during the COVID-19 Pandemic: Retrospective Descriptive Study in Thailand. Adv. Prev. Med. 2021, 2021, 5807056. [Google Scholar] [CrossRef]
- Puenpa, J.; Suwannakarn, K.; Chansaenroj, J.; Nilyanimit, P.; Yorsaeng, R.; Auphimai, C.; Kitphati, R.; Mungaomklang, A.; Kongklieng, A.; Chirathaworn, C.; et al. Molecular epidemiology of the first wave of severe acute respiratory syndrome coronavirus 2 infection in Thailand in 2020. Sci. Rep. 2020, 10, 16602. [Google Scholar] [CrossRef]
- Buathong, R.; Chaifoo, W.; Iamsirithaworn, S.; Wacharapluesadee, S.; Joyjinda, Y.; Rodpan, A.; Ampoot, W.; Putcharoen, O.; Paitoonpong, L.; Suwanpimolkul, G.; et al. Multiple clades of SARS-CoV-2 were introduced to Thailand during the first quarter of 2020. Microbiol. Immunol. 2021, 65, 405–409. [Google Scholar] [CrossRef]
- Rajatanavin, N.; Tuangratananon, T.; Suphanchaimat, R.; Tangcharoensathien, V. Responding to the COVID-19 second wave in Thailand by diversifying and adapting lessons from the first wave. BMJ Glob. Health 2021, 6, e006178. [Google Scholar] [CrossRef]
- Chookajorn, T.; Kochakarn, T.; Wilasang, C.; Kotanan, N.; Modchang, C. Southeast Asia is an emerging hotspot for COVID-19. Nat. Med. 2021, 27, 1495–1496. [Google Scholar] [CrossRef] [PubMed]
- Meo, S.A.; Meo, A.S.; Al-Jassir, F.F.; Klonoff, D.C. Omicron SARS-CoV-2 new variant: Global prevalence and biological and clinical characteristics. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 8012–8018. [Google Scholar] [CrossRef] [PubMed]
- Report of the WHO-China Joint Mission on Coronavirus Disease 2019 (COVID-19). Available online: https://www.who.int/publications/i/item/report-of-the-who-china-joint-mission-on-coronavirus-disease-2019-(covid-19) (accessed on 16 March 2022).
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Klempt, P.; Brzon, O.; Kasny, M.; Kvapilova, K.; Hubacek, P.; Briksi, A.; Bezdicek, M.; Koudelakova, V.; Lengerova, M.; Hajduch, M.; et al. Distribution of SARS-CoV-2 Lineages in the Czech Republic, Analysis of Data from the First Year of the Pandemic. Microorganisms 2021, 9, 1671. [Google Scholar] [CrossRef]
- Nagy, A.; Pongor, S.; Gyorffy, B. Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int. J. Antimicrob. Agents 2021, 57, 106272. [Google Scholar] [CrossRef] [PubMed]
- Aiewsakun, P.; Nilplub, P.; Wongtrakoongate, P.; Hongeng, S.; Thitithanyanont, A. SARS-CoV-2 genetic variations associated with COVID-19 pathogenicity. Microb. Genom. 2021, 7, 000734. [Google Scholar] [CrossRef]
- Oulas, A.; Zanti, M.; Tomazou, M.; Zachariou, M.; Minadakis, G.; Bourdakou, M.M.; Pavlidis, P.; Spyrou, G.M. Generalized linear models provide a measure of virulence for specific mutations in SARS-CoV-2 strains. PLoS ONE 2021, 16, e0238665. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Hui, K.P.Y.; Gu, H.; Ko, R.L.W.; Krishnan, P.; Ng, D.Y.M.; Liu, G.Y.Z.; Wan, C.K.C.; Cheung, M.C.; Ng, K.C.; et al. Introduction of ORF3a-Q57H SARS-CoV-2 Variant Causing Fourth Epidemic Wave of COVID-19, Hong Kong, China. Emerg. Infect. Dis. 2021, 27, 1492–1495. [Google Scholar] [CrossRef]
- Xu, H.; Xie, C.Y.; Li, P.H.; Ji, Z.L.; Sun, J.F.; Hu, B.; Li, X.; Fang, M. Demographic, Virological Characteristics and Prognosis of Asymptomatic COVID-19 Patients in South China. Front. Med. 2022, 9, 830942. [Google Scholar] [CrossRef]
- Pang, X.; Li, P.; Zhang, L.; Que, L.; Dong, M.; Xie, B.; Wang, Q.; Wei, Y.; Xie, X.; Li, L.; et al. Emerging Severe Acute Respiratory Syndrome Coronavirus 2 Mutation Hotspots Associated with Clinical Outcomes and Transmission. Front. Microbiol. 2021, 12, 753823. [Google Scholar] [CrossRef]
- Majumdar, P.; Niyogi, S. SARS-CoV-2 mutations: The biological trackway towards viral fitness. Epidemiol. Infect. 2021, 149, e110. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Haider, S.; Verma, M.; Singhvi, N.; Ponnusamy, K.; Malik, M.Z.; Verma, H.; Kumar, R.; Sood, U.; Hira, P.; et al. Comparative Genomics and Integrated Network Approach Unveiled Undirected Phylogeny Patterns, Co-mutational Hot Spots, Functional Cross Talk, and Regulatory Interactions in SARS-CoV-2. mSystems 2021, 6, e00030-21. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, A.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef]
- Ilmjarv, S.; Abdul, F.; Acosta-Gutierrez, S.; Estarellas, C.; Galdadas, I.; Casimir, M.; Alessandrini, M.; Gervasio, F.L.; Krause, K.H. Concurrent mutations in RNA-dependent RNA polymerase and spike protein emerged as the epidemiologically most successful SARS-CoV-2 variant. Sci. Rep. 2021, 11, 13705. [Google Scholar] [CrossRef]
- Garcia Borrega, J.; Naendrup, J.H.; Heindel, K.; Hamacher, L.; Heger, E.; Di Cristanziano, V.; Deppe, A.C.; Dusse, F.; Wetsch, W.A.; Eichenauer, D.A.; et al. Clinical Course and Outcome of Patients with SARS-CoV-2 Alpha Variant Infection Compared to Patients with SARS-CoV-2 Wild-Type Infection Admitted to the ICU. Microorganisms 2021, 9, 1944. [Google Scholar] [CrossRef]
- Grint, D.J.; Wing, K.; Houlihan, C.; Gibbs, H.P.; Evans, S.J.W.; Williamson, E.; McDonald, H.I.; Bhaskaran, K.; Evans, D.; Walker, A.J.; et al. Severity of Severe Acute Respiratory System Coronavirus 2 (SARS-CoV-2) Alpha Variant (B.1.1.7) in England. Clin. Infect. Dis. 2022, 75, e1120–e1127. [Google Scholar] [CrossRef]
- Florensa, D.; Mateo, J.; Spaimoc, R.; Miret, C.; Godoy, S.; Solsona, F.; Godoy, P. Severity of COVID-19 cases in the months of predominance of the Alpha and Delta variants. Sci. Rep. 2022, 12, 15456. [Google Scholar] [CrossRef]
- Snell, L.B.; Wang, W.; Alcolea-Medina, A.; Charalampous, T.; Batra, R.; de Jongh, L.; Higgins, F.; Nebbia, G.; Investigators, C.-U.H.; Wang, Y.; et al. Descriptive comparison of admission characteristics between pandemic waves and multivariable analysis of the association of the Alpha variant (B.1.1.7 lineage) of SARS-CoV-2 with disease severity in inner London. BMJ Open 2022, 12, e055474. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Della Ventura, C.; Menzo, S.; Bruzzone, B.; Sagradi, F.; Ceccherini-Silberstein, F.; Weisz, A.; Clementi, N.; Brindicci, G.; et al. Epidemiological and Clinical Features of SARS-CoV-2 Variants Circulating between April-December 2021 in Italy. Viruses 2022, 14, 2508. [Google Scholar] [CrossRef]
- Hill, V.; Du Plessis, L.; Peacock, T.P.; Aggarwal, D.; Colquhoun, R.; Carabelli, A.M.; Ellaby, N.; Gallagher, E.; Groves, N.; Jackson, B.; et al. The origins and molecular evolution of SARS-CoV-2 lineage B.1.1.7 in the UK. Virus Evol. 2022, 8, veac080. [Google Scholar] [CrossRef]
- Kurpas, M.K.; Jaksik, R.; Kus, P.; Kimmel, M. Genomic Analysis of SARS-CoV-2 Alpha, Beta and Delta Variants of Concern Uncovers Signatures of Neutral and Non-Neutral Evolution. Viruses 2022, 14, 2375. [Google Scholar] [CrossRef]
- GISAID. Tracking of hCoV-19 Variants. Available online: https://gisaid.org/hcov19-variants/ (accessed on 16 December 2021).
- Rahman, M.S.; Hoque, M.N.; Islam, M.R.; Islam, I.; Mishu, I.D.; Rahaman, M.M.; Sultana, M.; Hossain, M.A. Mutational insights into the envelope protein of SARS-CoV-2. Gene Rep. 2021, 22, 100997. [Google Scholar] [CrossRef]
- Low, Z.Y.; Zabidi, N.Z.; Yip, A.J.W.; Puniyamurti, A.; Chow, V.T.K.; Lal, S.K. SARS-CoV-2 Non-Structural Proteins and Their Roles in Host Immune Evasion. Viruses 2022, 14, 1991. [Google Scholar] [CrossRef]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: Open the door for novel therapies. Signal. Transduct. Target. Ther. 2022, 7, 26. [Google Scholar] [CrossRef]
- Ichikawa, T.; Torii, S.; Suzuki, H.; Takada, A.; Suzuki, S.; Nakajima, M.; Tampo, A.; Kakinoki, Y. Mutations in the nonstructural proteins of SARS-CoV-2 may contribute to adverse clinical outcome in patients with COVID-19. Int. J. Infect. Dis. 2022, 122, 123–129. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wave of Infections | n | Samples | Lineages (n) |
|---|---|---|---|
| First wave | 8 | VJR004, VJR007, VJR009, VJR010 | A.6 (4) |
| VJR008 | B (1) | ||
| VJR003, VJR005 | B.1 (2) | ||
| VJR002 | B.1.375 (1) | ||
| Second wave | 10 | VJR022, VJR023, VJR024, VJR025, VJR026, VJR027, VJR028, VJR029, VJR030, VJR031 | B.1.36.16 (10) |
| Third wave | 15 | VJR034, VJR037, VJR038, VJR039, VJR040, VJR041, VJR042, VJR043, VJR044, VJR045, VJR046, VJR047, VJR048, VJR050 | B.1.1.7 (14) |
| VJR035 | Q.3 (1) | ||
| Total | 33 | ||
| Total Cases (n = 32) | First Wave (n = 7) | Second Wave (n = 10) | Third Wave (n = 15) | |
|---|---|---|---|---|
| Age (Years) | ||||
| Median (range) | 37 (1–74) | 32 (13–48) | 34 (18–67) | 55 (1–74) |
| Gender, n (%) | ||||
| Male | 15 (46.9) | 3 (42.9) | 2 (20) | 10 (66.7) |
| Female | 17 (53.1) | 4 (57.1) | 8 (80) | 5 (33.3) |
| Symptoms, n (%) * | ||||
| Fever | 15 (57.7) | 2 (50) | 4 (50) | 9 (64.3) |
| Cough | 16 (61.5) | 2 (50) | 3 (37.5) | 11 (78.6) |
| Sore throat | 5 (19.2) | 2 (50) | 2 (25) | 1 (7.1) |
| Sputum production | 8 (30.8) | - | 2 (25) | 6 (42.9) |
| Headache | 3 (11.5) | 1 (25) | 1 (12.5) | 1 (7.1) |
| Nasal congestion | 4 (15.4) | - | 2 (25) | 2 (14.3) |
| Loss of taste and smell | 2 (7.7) | - | 1 (12.5) | 1 (7.1) |
| Fatigue | 4 (15.4) | - | 1 (12.5) | 3 (21.4) |
| Myalgia | 5 (19.2) | - | 2 (25) | 3 (21.4) |
| Breathing difficulty | 13 (50) | - | 2 (25) | 11 (78.6) |
| Diarrhea | 3 (11.5) | 1 (25) | - | 2 (14.3) |
| Other | 1 (3.8) | - | - | 1 (7.1) |
| COVID-19 disease severity, n (%) | ||||
| Asymptomatic | 6 (18.8) | 3 (42.9) | 2 (20) | 1 (6.7) |
| Mild | 13 (40.6) | 4 (57.1) | 6 (60) | 3 (20) |
| Moderate | 6 (18.8) | - | - | 6 (40) |
| Severe to critical | 7 (21.9) | - | 2 (20) | 5 (33.3) |
| Hospitalization | ||||
| No | 13 (40.6) | 7 (100) | 6 (60) | 0 |
| Yes | 19 (59.4) | 0 | 4 (40) | 15 (100) |
| Comorbidities | ||||
| Any *, n (%) | 11 (34.4) | - | 1 (10) | 10 (66.7) |
| Diabetes | 4 (12.5) | - | 1 (10) | 3 (20) |
| Hypertension | 8 (25.0) | - | 1 (10) | 7 (46.7) |
| Obesity | 1 (3.1) | - | - | 1 (6.7) |
| Cancer | 1 (3.1) | - | - | 1 (6.7) |
| Cerebrovascular disease | 2 (6.3) | - | - | 2 (13.3) |
| Heart disease | 3 (9.4) | - | - | 3 (20) |
| Final clinical outcomes, n (%) | ||||
| Recovered | 28 (87.5) | 7 (100) | 10 (100) | 11 (73.3) |
| Deceased | 4 (12.5) | - | - | 4 (26.7) |
| First Wave | Second Wave | Third Wave | |||||||
|---|---|---|---|---|---|---|---|---|---|
| No Symptom | Mild | No Symptom | Mild | Severe/ Critical | No Symptom | Mild | Moderate | Severe/ Critical | |
| Structural Protein | |||||||||
| Spike | A829T | A829T | L5F | S459F | L5F | H69del | H69del | V6F | H69del |
| D614G | S459F | D614G | S459F | V70del | V70del | H69del | V70del | ||
| D614G | D614G | Y144del | Y144del | V70del | Y144del | ||||
| N501Y | Y144F | Y144del | N501Y | ||||||
| A570D | N501Y | N501Y | T549I | ||||||
| D614G | A570D | A570D | A570D | ||||||
| P681H | D614G | D614G | D614G | ||||||
| T716I | P681H | P681H | P681H | ||||||
| S892A | T716I | T716I | T716I | ||||||
| D1118H | S892A | S892A | S892A | ||||||
| D1118H | D1118H | D1118H | |||||||
| Q784S | |||||||||
| Q787H | |||||||||
| I788V | |||||||||
| Envelope | S55F | ||||||||
| Nucleocapsid | S187L | S194L | S194L | S194L | D3L | D3L | D3L | D3L | |
| D402Y | R203K | R203K | R203K | R203K | |||||
| G204R | G204R | G204R | G204R | ||||||
| S235F | S235F | S235F | S235F | ||||||
| 247–251 del | |||||||||
| Non-structural protein | |||||||||
| Nsp1 | L16V | ||||||||
| Nsp2 | A476V | E373R | G47S | ||||||
| Nsp3 | L557F | L557F | D164G | D164G | P1200S | T183I | T183I | T183I | T183I |
| T583I | P1200S | P1200S | A890D | A890D | A890D | A890D | |||
| T860del | L1195I | I1412T | I1412T | I1412T | I1412T | ||||
| A861R | V393F | M1529I | Q1530H | D1755N | |||||
| L862M | T943N | Q1530E | A894del | ||||||
| F1110I | F1532V | L895V | |||||||
| Nsp6 | L37F | L37F | L37F | L37F | L37F | S106del | S106del | S106del | S106del |
| Q160K | G107del | G107del | G107del | G107del | |||||
| F108del | F108del | F108del | F108del | ||||||
| A54del | |||||||||
| Nsp9 | T109I | ||||||||
| Nsp12 | P323L | P323L | P323L | P323L | P323L | P323L | P323L | P323L | |
| Nsp13 | T588I | T588I | T588I | W167M | W167M | ||||
| A18V | |||||||||
| V232I | |||||||||
| Nsp14 | T215S | T215S | W227R | S369F | |||||
| T215R | 217–226 del | ||||||||
| 216–227 del | |||||||||
| Nsp15 | V172L | V172L | D282N | ||||||
| Nsp16 | D114del | D293G | |||||||
| V294I | |||||||||
| NS3 | G251V | Q57H | Q57H | Q57H | G254stop | G254stop | G254stop | G254stop | |
| Q57H | G254stop | I232L | |||||||
| S216P | V55F | ||||||||
| V13L | D27Y | ||||||||
| I232L | |||||||||
| NS7b | C41F | ||||||||
| NS8 | L84S | L84S | Q27stop | Q27stop | Q27stop | Q27stop | |||
| D75Y | R52I | R52I | R52I | R52I | |||||
| K68stop | K68stop | K68stop | K68stop | ||||||
| Y73C | Y73C | Y73C | Y73C | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Setthapramote, C.; Wongsuk, T.; Thongnak, C.; Phumisantiphong, U.; Hansirisathit, T.; Thanunchai, M. SARS-CoV-2 Variants by Whole-Genome Sequencing in a University Hospital in Bangkok: First to Third COVID-19 Waves. Pathogens 2023, 12, 626. https://doi.org/10.3390/pathogens12040626
Setthapramote C, Wongsuk T, Thongnak C, Phumisantiphong U, Hansirisathit T, Thanunchai M. SARS-CoV-2 Variants by Whole-Genome Sequencing in a University Hospital in Bangkok: First to Third COVID-19 Waves. Pathogens. 2023; 12(4):626. https://doi.org/10.3390/pathogens12040626
Chicago/Turabian StyleSetthapramote, Chayanee, Thanwa Wongsuk, Chuphong Thongnak, Uraporn Phumisantiphong, Tonsan Hansirisathit, and Maytawan Thanunchai. 2023. "SARS-CoV-2 Variants by Whole-Genome Sequencing in a University Hospital in Bangkok: First to Third COVID-19 Waves" Pathogens 12, no. 4: 626. https://doi.org/10.3390/pathogens12040626