

Rapid Identification of ASFV, CSFV and FMDV from Mongolian Outbreaks with MinION Short Amplicon Sequencing
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Extraction of Nucleic Acids
2.2. Polymerase Chain Reaction (PCR) and Reverse-Transcription (RT-) PCR
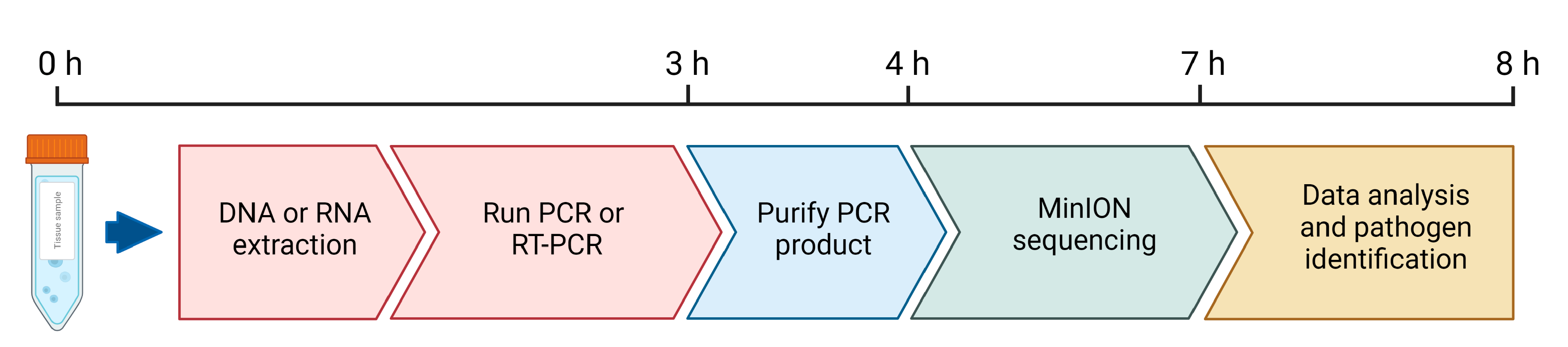
2.3. Oxford Nanopore MinION Sequencing
2.4. Sequencing Data Processing and Analysis
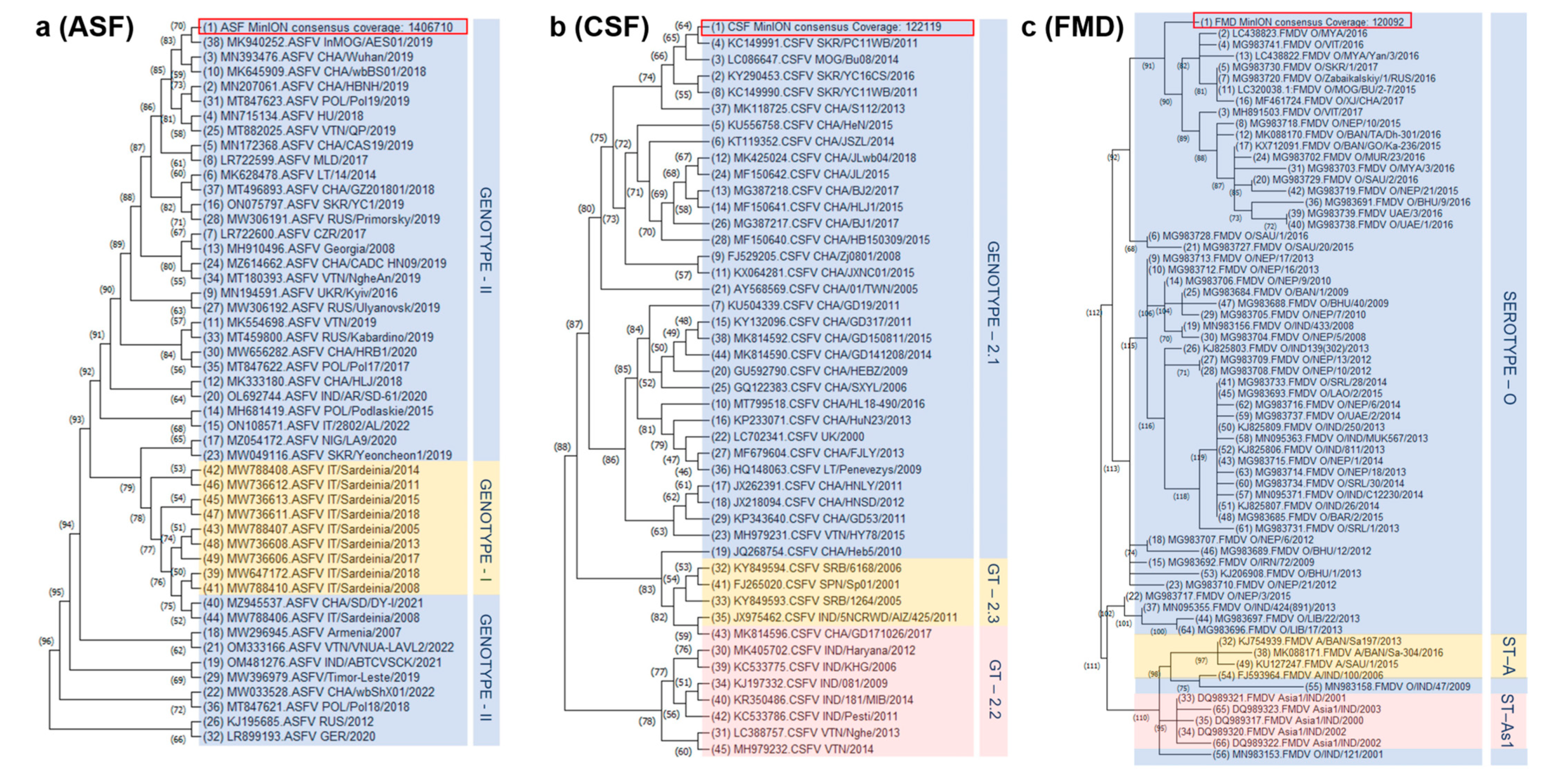
2.5. Sequence Alignment and Phylogenetic Analysis
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WOAH. Foot and Mouth Disease; WOAH: France, 2022. [Google Scholar]
- Clemmons, E.A.; Alfson, K.J.; Dutton, J.W.,3rd. Transboundary Animal Diseases, an Overview of 17 Diseases with Potential for Global Spread and Serious Consequences. Animals 2021, 11, 2039. [Google Scholar] [CrossRef]
- WRLFMD. FMD Reports of the World Reference Laboratory; WRLFMD: UK, 2022. [Google Scholar]
- Ankhanbaatar, U.; Sainnokhoi, T.; Khanui, B.; Ulziibat, G.; Jargalsaikhan, T.; Purevtseren, D.; Settypalli, T.B.K.; Flannery, J.; Dundon, W.G.; Basan, G.; et al. African swine fever virus genotype II in Mongolia, 2019. Transbound. Emerg. Dis. 2021, 68, 2787–2794. [Google Scholar] [CrossRef] [PubMed]
- Enkhbold, B.; Shatar, M.; Wakamori, S.; Tamura, T.; Hiono, T.; Matsuno, K.; Okamatsu, M.; Umemura, T.; Damdinjav, B.; Sakoda, Y. Genetic and virulence characterization of classical swine fever viruses isolated in Mongolia from 2007 to 2015. Virus Genes. 2017, 53, 418–425. [Google Scholar] [CrossRef]
- Ulziibat, G.; Myagmarsuren, O.; Khisgee Basan, G.; Sandag, B.; Ruuragch, S.; Limon, G.; Wilsden, G.; Browning, C.; King, D.P. Immunogenicity of imported foot-and-mouth vaccines in different species in Mongolia. Vaccine 2020, 38, 1708–1714. [Google Scholar] [CrossRef]
- Gaudreault, N.N.; Madden, D.W.; Wilson, W.C.; Trujillo, J.D.; Richt, J.A. African Swine Fever Virus: An Emerging DNA Arbovirus. Front. Vet. Sci. 2020, 7, 215. [Google Scholar] [CrossRef] [PubMed]
- Cwynar, P.; Stojkov, J.; Wlazlak, K. African Swine Fever Status in Europe. Viruses 2019, 11, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazur-Panasiuk, N.; Zmudzki, J.; Wozniakowski, G. African Swine Fever Virus—Persistence in Different Environmental Conditions and the Possibility of its Indirect Transmission. J. Vet. Res. 2019, 63, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- USDA. APHIS Adds the Dominican Republic to the List of Regions Affected with African Swine Fever. 2021. Available online: https://www.aphis.usda.gov/aphis/newsroom/federal-register-posts/sa_by_date/sa_2021/asf-dominican-republic (accessed on 1 February 2023).
- Ito, S.; Bosch, J.; Martinez-Alives, M.; Sanchez-Vizcaino, J.M. The Evolution of African Swine Fever in China: A Global Threat? Front. Vet. Sci. 2022, 9, 828498. [Google Scholar] [CrossRef]
- Le, V.P.; Jeong, D.G.; Yoon, S.W.; Kwon, H.M.; Trinh, T.B.N.; Nguyen, T.L.; Bui, T.T.N.; Oh, J.; Kim, J.B.; Cheong, K.M.; et al. Outbreak of African Swine Fever, Vietnam, 2019. Emerg. Infect. Dis. 2019, 25, 1433–1435. [Google Scholar] [CrossRef]
- Rowlands, R.J.; Michaud, V.; Heath, L.; Hutchings, G.; Oura, C.; Vosloo, W.; Dwarka, R.; Onashvili, T.; Albina, E.; Dixo, L.K. African swine fever virus isolate, Georgia, 2007. Emerg. Infect. Dis. 2008, 14, 1870–1874. [Google Scholar] [CrossRef]
- Shao, Q.; Li, R.; Han, Y.; Han, D.; Qiu, J. Temporal and Spatial Evolution of the African Swine Fever Epidemic in Vietnam. Int. J. Environ. Res. Public. Health 2022, 19, 8001. [Google Scholar] [CrossRef] [PubMed]
- Szymanska, E.J.; Dziwulaki, M. Development of African Swine Fever in Poland. Agriculture 2022, 12, 119. [Google Scholar] [CrossRef]
- Vilem, A.; Nurmoja, I.; Niine, T.; Riit, T.; Nieto, R.; Viltrop, A.; Gallardo, C. Molecular Characterization of African Swine Fever Virus Isolates in Estonia in 2014–2019. Pathogens 2020, 9, 582. [Google Scholar] [CrossRef]
- Edwards, S.; Fukusho, A.; Lefevre, P.C.; Lipowski, A.; Pejsak, Z.; Roehe, P.; Westergaard, J. Classical swine fever: The global situation. Vet. Microbiol. 2000, 73, 103–119. [Google Scholar] [CrossRef]
- Wei, Q.; Liu, Y.; Zhang, G. Research Progress and Challenges in Vaccine Development against Classical Swine Fever Virus. Viruses 2021, 13, 445. [Google Scholar] [CrossRef] [PubMed]
- Mahy, B.W. Introduction and history of foot-and-mouth disease virus. Curr. Top. Microbiol. Immunol. 2005, 288, 1–8. [Google Scholar]
- Knight-Jones, T.J.; Rushton, J. The economic impacts of foot and mouth disease—What are they, how big are they and where do they occur? Prev. Vet. Med. 2013, 112, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Torres-Velez, F.; Havas, K.A.; Spiegel, K.; Brown, C. Transboundary animal diseases as re-emerging threats—Impact on one health. Semin. Diagn. Pathol. 2019, 36, 193–196. [Google Scholar] [CrossRef]
- WOAH. Terrestrial Animal Health Code; WOAH: France, 2021. [Google Scholar]
- Eberling, A.J.; Bieker-Stefanelli, J.; Reising, M.M.; Siev, D.; Martin, B.M.; Mclntosh, M.T.; Beckam, T.R. Development, optimization, and validation of a Classical swine fever virus real-time reverse transcription polymerase chain reaction assay. J. Vet. Diagn. Investig. 2011, 23, 994–998. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.X.; Shi, K.C.; Zhao, J.; Yin, Y.W.; Si, H.B.; Qu, S.J.; Lu, W.J.; Chen, Y.; Long, F. Development of a one-step multiplex qRT-PCR assay for the detection of African swine fever virus, classical swine fever virus and atypical porcine pestivirus. Bmc Vet. Res. 2022, 18, 43. [Google Scholar] [CrossRef]
- Risatti, G.R.; Callahan, J.D.; Nelson, W.M.; Borca, M.V. Rapid detection of classical swine fever virus by a portable real-time reverse transcriptase PCR assay. J. Clin. Microbiol. 2003, 41, 500–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, E.C.; Colling, G.; Gurung, R.B.; Allen, J. Thepotential of diagnostic point-of-care tests (POCTs) for infectious and zoonotic animal diseases in developing countries: Technical, regulatory and sociocultural considerations. Transbound. Emerg. Dis. 2021, 68, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Howson, E.L.A.; Armson, B.; Lyons, N.A.; Lyons, N.A.; Chepkwony, E.; Kasanga, C.J.; Kandusi, S.; Ndusilo, N.; Ymazaki, W.; Gizaw, D.; et al. Direct detection and characterization of foot-and-mouth disease virus in East Africa using a field-ready real-time PCR platform. Transbound. Em, erg. Dis. 2018, 65, 221–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight-Jones, T.J.; Robinson, L.; Charleston, B.; Rodriguez, L.L.; Gay, C.G.; Sumpton, K.J.; Vosloo, W. Global Foot-and-Mouth Disease Research Update and Gap Analysis: 4—Diagnostics. Transbound. Emerg. Dis. 2016, 63 (Suppl. S1), 42–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanopore, O. What’s in my Pot? (WIMP), A Quantitative Analysis Tool for Real-Time Species Identification; Oxford Nanopore Technologies: UK, 2017. [Google Scholar]
- McDowell, C.D.; Bold, D.; Trujillo, J.D.; Meekins, D.A.; Keating, C.; Cool, K.; Kwon, T.; Madden, D.W.; Artiaga, B.L.; Balaraman, V.; et al. Experimental Infection of Domestic Pigs with African Swine Fever Virus Isolated in 2019 in Mongolia. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Van Borm, S.; Wang, J.; Granberg, F.; Colling, A. Next-generation sequencing workflows in veterinary infection biology: Towards validation and quality assurance. Rev. Sci. Tech. 2016, 35, 67–81. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, L.G.; Mechler-Dreibi, M.L.; Almeda, H.M.S.; Gatto, I.R.H. Bovine Viral Diarrhea Virus: Recent Findings about Its Occurrence in Pigs. Viruses 2020, 12, 600. [Google Scholar] [CrossRef]
- Tao, J.; Liao, J.; Wang, Y.; Zhang, X.; Wang, J.; Zhu, G. Bovine viral diarrhea virus (BVDV) infections in pigs. Vet. Microbiol. 2013, 165, 185–189. [Google Scholar] [CrossRef]
- Sammin, D.; Ryan, E.; Ferris, N.P.; King, D.P.; Zientara, S.; Haas, B.; Yadin, H.; Alexandersen, S.; Sumpton, K.; Paton, D.J. Options for decentralized testing of suspected secondary outbreaks of foot-and-mouth disease. Transbound. Emerg. Dis. 2010, 57, 237–243. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
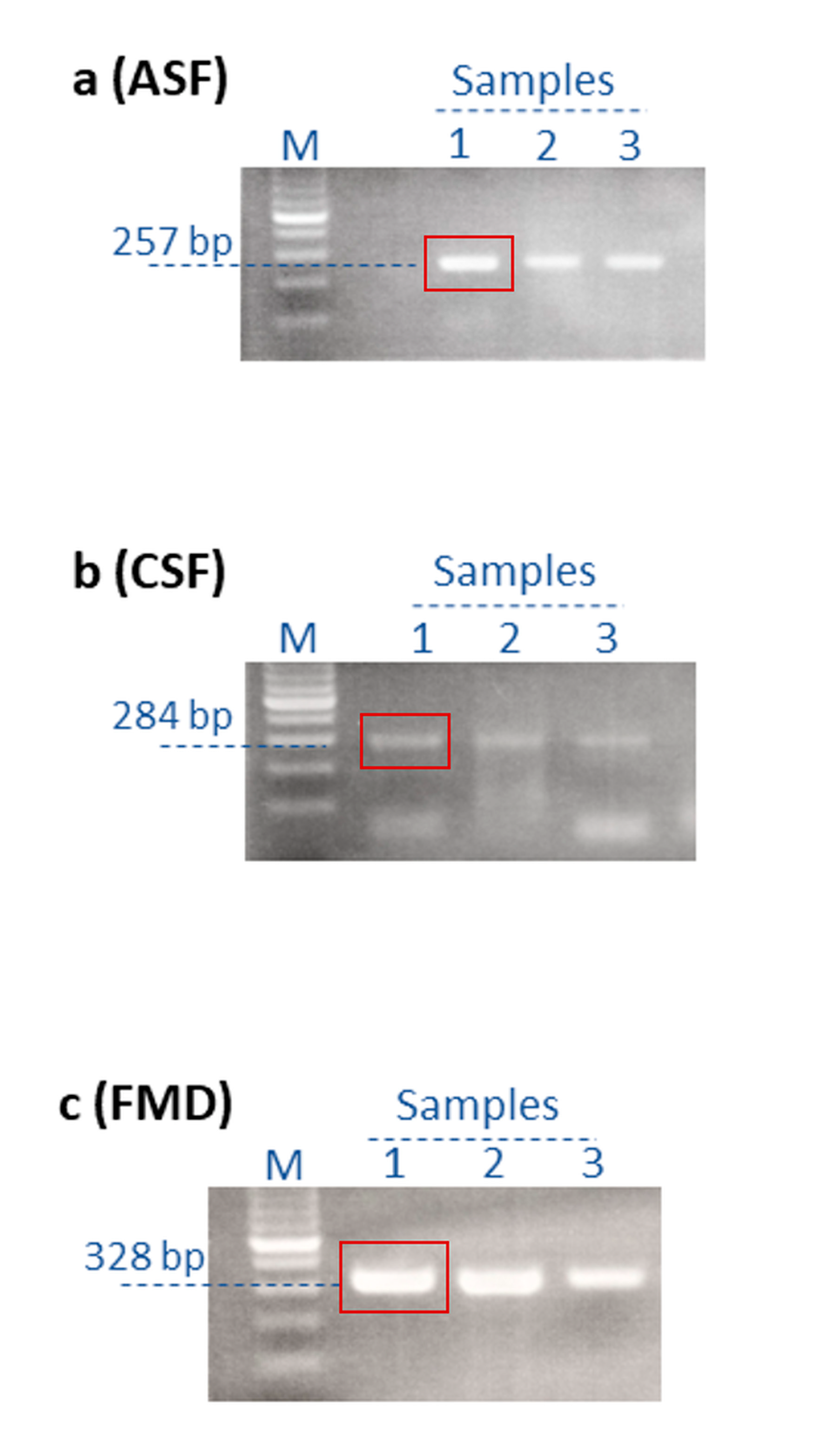
| Pathogen | Virus Geno/Serotype | Tissue | Collection Date | Sample Origin | Animal Species |
|---|---|---|---|---|---|
| ASFV | Genotype II | Spleen | January 2019 | Bulgan province | Domestic pig |
| CSFV | Genotype 2.1b | Blood | April 2015 | Khan-Uul District Ulaanbaatar | Domestic pig |
| FMDV | Serotype O | Tongue epithelium | January 2019 | Dornod province | Domestic Cattle |
| Pathogen | Position | Forward Primer | Reverse Primer | Length |
|---|---|---|---|---|
| ASFV | p72 | 5′-AGTTATGGGAAACCCGACCC-3′ | 5′-CCCTGAATCGGAGCATCCT-3′ | 257 bp |
| CSFV | IRES | 5’-ATGCCCACAGTAGGACTAGCA-3′ | 5’-TCAACTCCATGTGCCATGTAC-3’ | 284 bp |
| FMDV | 5′ UTR | 5′-GCCTGGTCTTTCCAGGTCT-3′ | 5′-CCAGTCCCCTTCTCAGATC-3′ | 328 bp |
| Row Labels | Count of Reads | Average of Score |
|---|---|---|
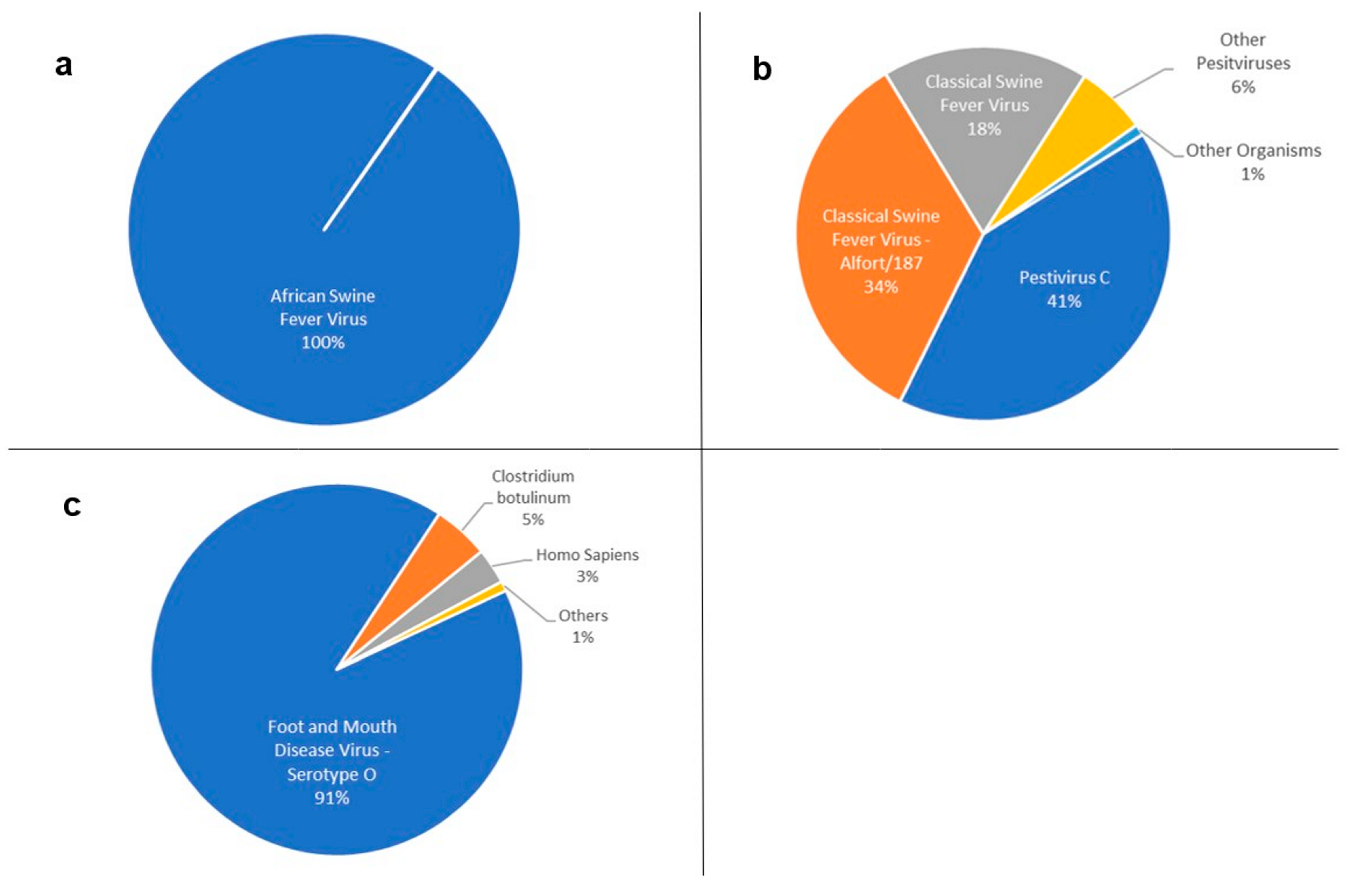
| Pestivirus C | 42,309 | 1010.36 |
| Classical swine fever virus—Alfort/187 | 36,259 | 997.24 |
| Classical swine fever virus | 18,808 | 1087.15 |
| Pestivirus | 4612 | 312.34 |
| Aydin-like pestivirus | 518 | 241.08 |
| Bovine viral diarrhea virus 1 | 242 | 430.90 |
| Pestivirus giraffe-1 H138 | 77 | 336.71 |
| Border disease virus | 20 | 392.25 |
| Bovine viral diarrhea virus 2 | 7 | 165.57 |
| Bovine viral diarrhea virus 3 Th/04_KhonKaen | 7 | 179.29 |
| Pronghorn antelope pestivirus | 1 | 169.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bold, D.; Souza-Neto, J.A.; Gombo-Ochir, D.; Gaudreault, N.N.; Meekins, D.A.; McDowell, C.D.; Zayat, B.; Richt, J.A. Rapid Identification of ASFV, CSFV and FMDV from Mongolian Outbreaks with MinION Short Amplicon Sequencing. Pathogens 2023, 12, 533. https://doi.org/10.3390/pathogens12040533
Bold D, Souza-Neto JA, Gombo-Ochir D, Gaudreault NN, Meekins DA, McDowell CD, Zayat B, Richt JA. Rapid Identification of ASFV, CSFV and FMDV from Mongolian Outbreaks with MinION Short Amplicon Sequencing. Pathogens. 2023; 12(4):533. https://doi.org/10.3390/pathogens12040533
Chicago/Turabian StyleBold, Dashzeveg, Jayme A. Souza-Neto, Delgerzul Gombo-Ochir, Natasha N. Gaudreault, David A. Meekins, Chester D. McDowell, Batsukh Zayat, and Juergen A. Richt. 2023. "Rapid Identification of ASFV, CSFV and FMDV from Mongolian Outbreaks with MinION Short Amplicon Sequencing" Pathogens 12, no. 4: 533. https://doi.org/10.3390/pathogens12040533