
Synergistic Detrimental Effects of Cigarette Smoke, Alcohol, and SARS-CoV-2 in COPD Bronchial Epithelial Cells
 , , and
, , and 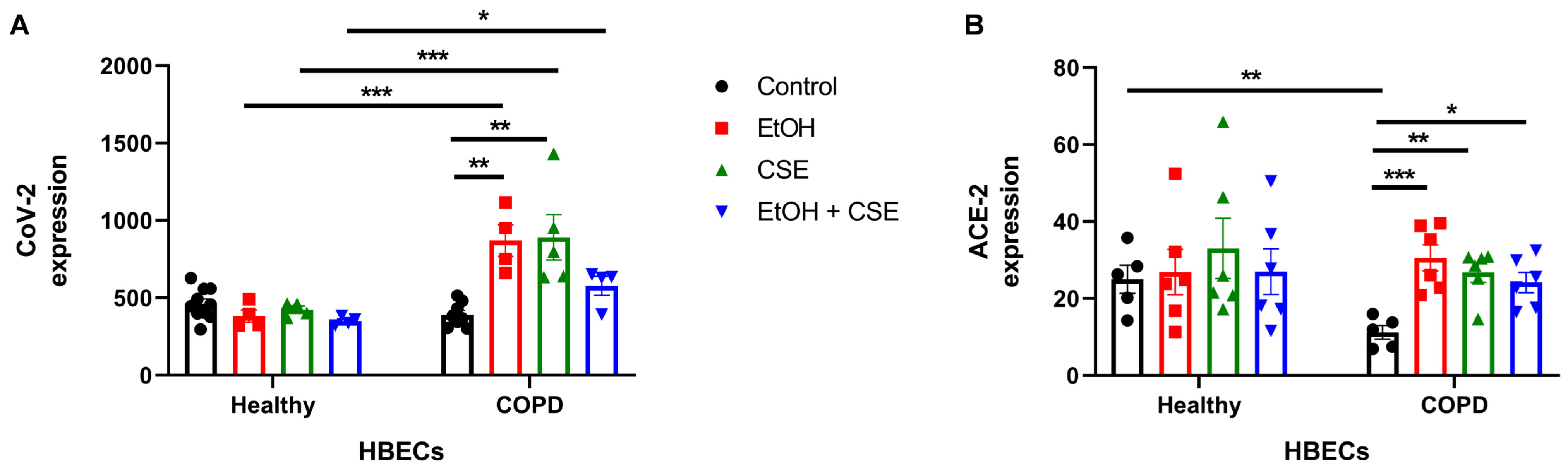{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Culture of Human Bronchial Epithelial Cells (HBEC)
2.2. Virus
2.3. HBEC Treatments and Infection
2.4. RNA Extraction and Quantitative Polymerase Chain Reaction (qPCR)
2.5. Lactate Dehydrogenase (LDH) Activity Assay
2.6. Interleukin-8 (IL-8) ELISA
2.7. Statistical Analysis
3. Results
3.1. Short Exposure to Cigarette Smoke or Alcohol Augments SARS-CoV-2 Infection in HBECs Isolated from COPD Patients
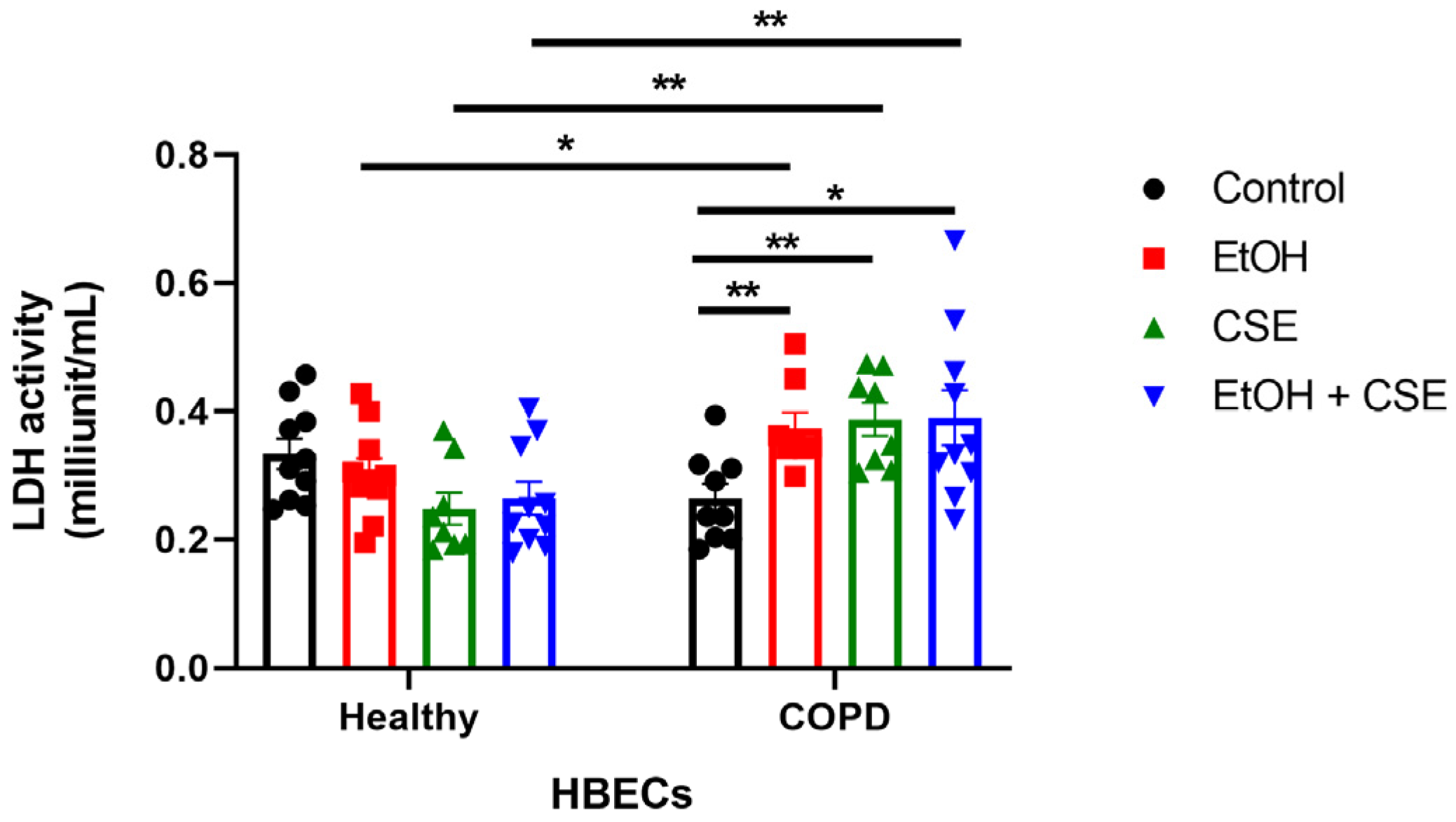
3.2. SARS-CoV-2 Infection following Short Exposure to Cigarette Smoke and Alcohol Exacerbates Injury in HBECs from COPD Patients but Not in Healthy Patients
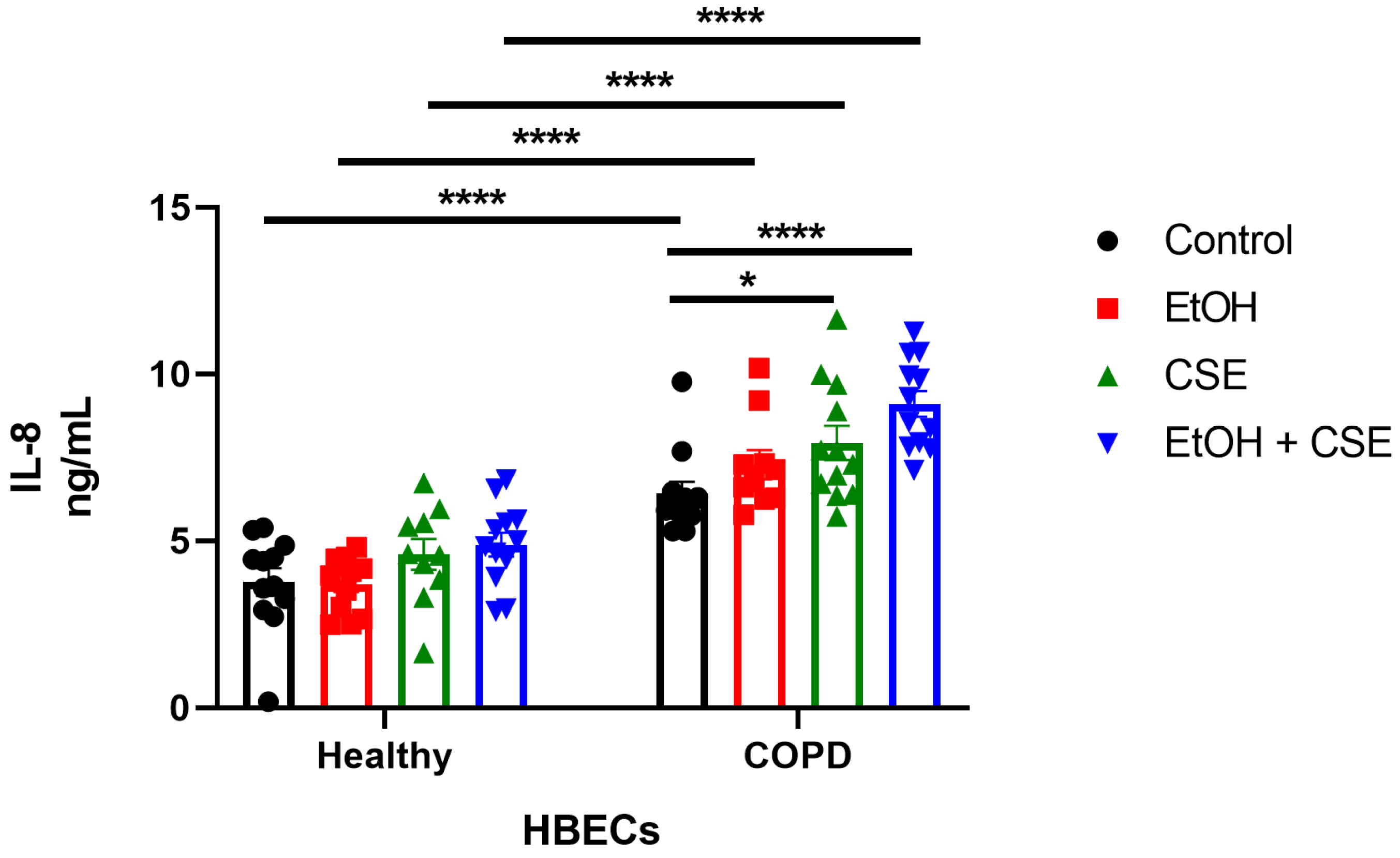
3.3. Elevated IL-8 Secretion Correlated to the Synergistic Damage Induced by SARS-CoV-2, Cigarette Smoke, and Alcohol in COPD HBECs
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Office on Smoking and Health, Centers for Disease Control and Prevention. Smoking & Tobacco Use: Fast Facts. 2022. Available online: https://www.cdc.gov/tobacco/data_statistics/fact_sheets/fast_facts/index.htm#beginning (accessed on 5 October 2022).
- Romberger, D.J.; Grant, K. Alcohol consumption and smoking status: The role of smoking cessation. Biomed. Pharmacother. 2004, 58, 77–83. [Google Scholar] [CrossRef]
- Gentry, M.J. Pneumococcal pneumonia in alcoholism and HIV infection. Alcohol Clin. Exp. Res. 1998, 22, 201s–203s. [Google Scholar] [CrossRef]
- Tabak, C.; Smit, H.A.; Rasanen, L.; Fidanza, F.; Menotti, A.; Nissinen, A.; Feskens, E.J.; Heederik, D.; Kromhout, D. Alcohol consumption in relation to 20-year COPD mortality and pulmonary function in middle-aged men from three European countries. Epidemiology 2001, 12, 239–245. [Google Scholar] [CrossRef]
- Yeligar, S.M.; Chen, M.M.; Kovacs, E.J.; Sisson, J.H.; Burnham, E.L.; Brown, L.A. Alcohol and lung injury and immunity. Alcohol 2016, 55, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Ganesh, B.; Rajakumar, T.; Malathi, M.; Manikandan, N.; Nagaraj, J.; Santhakumar, A.; Elangovan, A.; Malik, Y.S. Epidemiology and pathobiology of SARS-CoV-2 (COVID-19) in comparison with SARS, MERS: An updated overview of current knowledge and future perspectives. Clin. Epidemiol. Glob. Health 2021, 10, 100694. [Google Scholar] [CrossRef]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-l.; Hui, D.S. Clinical characteristics of coronavirus disease 2019 in China. New Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. Jama 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Wu, J.T.; Leung, K.; Bushman, M.; Kishore, N.; Niehus, R.; de Salazar, P.M.; Cowling, B.J.; Lipsitch, M.; Leung, G.M. Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan, China. Nat. Med. 2020, 26, 506–510. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, S.; Liu, J.; Zhang, Z.; Wan, X.; Huang, B.; Chen, Y.; Zhang, Y. COVID-19: Immunopathogenesis and Immunotherapeutics. Signal Transduct. Target. Ther. 2020, 5, 128. [Google Scholar] [CrossRef]
- Yi, Y.; Lagniton, P.N.; Ye, S.; Li, E.; Xu, R.-H. COVID-19: What has been learned and to be learned about the novel coronavirus disease. Int. J. Biol. Sci. 2020, 16, 1753. [Google Scholar] [CrossRef]
- Mueller, A.L.; McNamara, M.S.; Sinclair, D.A. Why does COVID-19 disproportionately affect older people? Aging 2020, 12, 9959. [Google Scholar] [CrossRef]
- Popkin, B.M.; Du, S.; Green, W.D.; Beck, M.A.; Algaith, T.; Herbst, C.H.; Alsukait, R.F.; Alluhidan, M.; Alazemi, N.; Shekar, M. Individuals with obesity and COVID-19: A global perspective on the epidemiology and biological relationships. Obes. Rev. 2020, 21, e13128. [Google Scholar] [CrossRef]
- Van Zyl-Smit, R.N.; Richards, G.; Leone, F.T. Tobacco smoking and COVID-19 infection. Lancet Respir. Med. 2020, 8, 664–665. [Google Scholar] [CrossRef]
- Bailey, K.L.; Sayles, H.; Campbell, J.; Khalid, N.; Anglim, M.; Ponce, J.; Wyatt, T.A.; McClay, J.C.; Burnham, E.L.; Anzalone, A.; et al. COVID-19 patients with documented alcohol use disorder or alcohol-related complications are more likely to be hospitalized and have higher all-cause mortality. Alcohol Clin. Exp. Res. 2022, 46, 1023–1035. [Google Scholar] [CrossRef]
- Pranata, R.; Soeroto, A.; Huang, I.; Lim, M.; Santoso, P.; Permana, H.; Lukito, A. Effect of chronic obstructive pulmonary disease and smoking on the outcome of COVID-19. Int. J. Tuberc. Lung Dis. 2020, 24, 838–843. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Alcohol Use and Your Health. Available online: https://www.cdc.gov/alcohol/fact-sheets/alcohol-use.htm (accessed on 8 August 2022).
- De Roux, A.; Cavalcanti, M.; Marcos, M.A.; Garcia, E.; Ewig, S.; Mensa, J.; Torres, A. Impact of Alcohol Abuse in the Etiology and Severity of Community-Acquired Pneumonia. Chest 2006, 129, 1219–1225. [Google Scholar] [CrossRef]
- Bailey, K.L.; Wyatt, T.A.; Katafiasz, D.M.; Taylor, K.W.; Heires, A.J.; Sisson, J.H.; Romberger, D.J.; Burnham, E.L. Alcohol and cannabis use alter pulmonary innate immunity. Alcohol 2019, 80, 131–138. [Google Scholar] [CrossRef]
- Gómez Antúnez, M.; Muiño Míguez, A.; Bendala Estrada, A.D.; Maestro de la Calle, G.; Monge Monge, D.; Boixeda, R.; Ena, J.; Mella Pérez, C.; Anton Santos, J.M.; Lumbreras Bermejo, C. Clinical Characteristics and Prognosis of COPD Patients Hospitalized with SARS-CoV-2. Int. J. Chronic Obs. Pulmon. Dis. 2020, 15, 3433–3445. [Google Scholar] [CrossRef]
- Fulcher, M.L.; Gabriel, S.; Burns, K.A.; Yankaskas, J.R.; Randell, S.H. Well-differentiated human airway epithelial cell cultures. In Human Cell Culture Protocols; Springer: Berlin/Heidelberg, Germany, 2005; pp. 183–206. [Google Scholar]
- Sisson, J.H.; Stoner, J.A.; Ammons, B.A.; Wyatt, T.A. All-digital image capture and whole-field analysis of ciliary beat frequency. J. Microsc. 2003, 211, 103–111. [Google Scholar] [CrossRef]
- Ochoa, C.A.; Nissen, C.G.; Mosley, D.D.; Bauer, C.D.; Jordan, D.L.; Bailey, K.L.; Wyatt, T.A. Aldehyde Trapping by ADX-102 Is Protective against Cigarette Smoke and Alcohol Mediated Lung Cell Injury. Biomolecules 2022, 12, 393. [Google Scholar] [CrossRef]
- Higham, A.; Bostock, D.; Booth, G.; Dungwa, J.V.; Singh, D. The effect of electronic cigarette and tobacco smoke exposure on COPD bronchial epithelial cell inflammatory responses. Int. J. Chronic. Obs. Pulmon. Dis. 2018, 13, 989–1000. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.; Rampersaud, R.; Aguilar, J.L.; Randis, T.M.; Kreindler, J.L.; Ratner, A.J. Cigarette smoke inhibits airway epithelial cell innate immune responses to bacteria. Infect. Immun. 2010, 78, 2146–2152. [Google Scholar] [CrossRef] [Green Version]
- Richter, A.; O’Donnell, R.A.; Powell, R.M.; Sanders, M.W.; Holgate, S.T.; Djukanovic, R.; Davies, D.E. Autocrine ligands for the epidermal growth factor receptor mediate interleukin-8 release from bronchial epithelial cells in response to cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2002, 27, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Schamberger, A.C.; Staab-Weijnitz, C.A.; Mise-Racek, N.; Eickelberg, O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci. Rep. 2015, 5, 8163. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, X.; Umino, T.; Skold, C.M.; Zhu, Y.; Kohyama, T.; Spurzem, J.R.; Romberger, D.J.; Rennard, S.I. Cigarette smoke inhibits human bronchial epithelial cell repair processes. Am. J. Respir. Cell Mol. Biol. 2001, 25, 772–779. [Google Scholar] [CrossRef]
- Jordan, D.; Mosley, D.; Ochoa, C.; Bauer, C.; Nissen, C.; Heires, A.J.; Romberger, D.J.; Wyatt, T.A. Alcohol and Cigarette Smoke Decrease Lung Epithelial Cell Proliferation; University of Nebraska Press: Lincoln, NE, USA, 2021. [Google Scholar]
- Simet, S.M.; Wyatt, T.A.; DeVasure, J.; Yanov, D.; Allen-Gipson, D.; Sisson, J.H. Alcohol increases the permeability of airway epithelial tight junctions in Beas-2B and NHBE cells. Alcohol Clin. Exp. Res. 2012, 36, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, T.A.; Bailey, K.L.; Simet, S.M.; Warren, K.J.; Sweeter, J.M.; DeVasure, J.M.; Pavlik, J.A.; Sisson, J.H. Alcohol potentiates RSV-mediated injury to ciliated airway epithelium. Alcohol 2019, 80, 17–24. [Google Scholar] [CrossRef]
- Wyatt, T.A.; Canady, K.; Heires, A.J.; Poole, J.A.; Bailey, K.L.; Nordgren, T.M.; Romberger, D.J. Alcohol inhibits organic dust-induced icam-1 expression on bronchial epithelial cells. Safety 2017, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, T.A.; Sisson, J.H. Chronic ethanol downregulates PKA activation and ciliary beating in bovine bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L575–L581. [Google Scholar] [CrossRef] [Green Version]
- Schulz, C.; Wolf, K.; Harth, M.; Kratzel, K.; Kunz-Schughart, L.; Pfeifer, M. Expression and release of interleukin-8 by human bronchial epithelial cells from patients with chronic obstructive pulmonary disease, smokers, and never-smokers. Respiration 2003, 70, 254–261. [Google Scholar] [CrossRef]
- Ganesan, S.; Unger, B.L.; Comstock, A.T.; Angel, K.A.; Mancuso, P.; Martinez, F.J.; Sajjan, U.S. Aberrantly activated EGFR contributes to enhanced IL-8 expression in COPD airways epithelial cells via regulation of nuclear FoxO3A. Thorax 2013, 68, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Hollander, C.; Sitkauskiene, B.; Sakalauskas, R.; Westin, U.; Janciauskiene, S.M. Serum and bronchial lavage fluid concentrations of IL-8, SLPI, sCD14 and sICAM-1 in patients with COPD and asthma. Respir. Med. 2007, 101, 1947–1953. [Google Scholar] [CrossRef] [Green Version]
- Hulina-Tomaskovic, A.; Heijink, I.H.; Jonker, M.R.; Somborac-Bacura, A.; Grdic Rajkovic, M.; Rumora, L. Pro-inflammatory effects of extracellular Hsp70 and cigarette smoke in primary airway epithelial cells from COPD patients. Biochimie 2019, 156, 47–58. [Google Scholar] [CrossRef]
- Osei, E.T.; Noordhoek, J.A.; Hackett, T.L.; Spanjer, A.I.; Postma, D.S.; Timens, W.; Brandsma, C.A.; Heijink, I.H. Interleukin-1alpha drives the dysfunctional cross-talk of the airway epithelium and lung fibroblasts in COPD. Eur. Respir. J. 2016, 48, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Patel, I.S.; Roberts, N.J.; Lloyd-Owen, S.J.; Sapsford, R.J.; Wedzicha, J.A. Airway epithelial inflammatory responses and clinical parameters in COPD. Eur. Respir. J. 2003, 22, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, C.; Yoneda, T.; Yoshikawa, M.; Fu, A.; Tokuyama, T.; Tsukaguchi, K.; Narita, N. Airway inflammation in COPD assessed by sputum levels of interleukin-8. Chest 1997, 112, 505–510. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zheng, H.; Zhang, H.; Ma, W.; Wang, F.; Liu, C.; He, S. Increased interleukin (IL)-8 and decreased IL-17 production in chronic obstructive pulmonary disease (COPD) provoked by cigarette smoke. Cytokine 2011, 56, 717–725. [Google Scholar] [CrossRef]
- Lee, K.H.; Lee, C.H.; Woo, J.; Jeong, J.; Jang, A.H.; Yoo, C.G. Cigarette Smoke Extract Enhances IL-17A-Induced IL-8 Production via Up-Regulation of IL-17R in Human Bronchial Epithelial Cells. Mol. Cells 2018, 41, 282–289. [Google Scholar] [CrossRef]
- Lee, K.H.; Lee, J.; Jeong, J.; Woo, J.; Lee, C.H.; Yoo, C.G. Cigarette smoke extract enhances neutrophil elastase-induced IL-8 production via proteinase-activated receptor-2 upregulation in human bronchial epithelial cells. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef]
- Mio, T.; Romberger, D.J.; Thompson, A.B.; Robbins, R.A.; Heires, A.; Rennard, S.I. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am. J. Respir. Crit. Care Med. 1997, 155, 1770–1776. [Google Scholar] [CrossRef]
- Moon, H.G.; Zheng, Y.; An, C.H.; Kim, Y.K.; Jin, Y. CCN1 secretion induced by cigarette smoking extracts augments IL-8 release from bronchial epithelial cells. PLoS ONE 2013, 8, e68199. [Google Scholar] [CrossRef]
- Hellermann, G.R.; Nagy, S.B.; Kong, X.; Lockey, R.F.; Mohapatra, S.S. Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cells. Respir. Res. 2002, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Kode, A.; Yang, S.R.; Rahman, I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir. Res. 2006, 7, 132. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.K.; Chan, S.C.; Law, A.C.; Ip, M.S.; Mak, J.C. The role of MAPK and Nrf2 pathways in ketanserin-elicited attenuation of cigarette smoke-induced IL-8 production in human bronchial epithelial cells. Toxicol. Sci. 2012, 125, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Moretto, N.; Bertolini, S.; Iadicicco, C.; Marchini, G.; Kaur, M.; Volpi, G.; Patacchini, R.; Singh, D.; Facchinetti, F. Cigarette smoke and its component acrolein augment IL-8/CXCL8 mRNA stability via p38 MAPK/MK2 signaling in human pulmonary cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L929–L938. [Google Scholar] [CrossRef]
- Mulligan, R.M.; Atkinson, C.; Vertegel, A.A.; Reukov, V.; Schlosser, R.J. Cigarette smoke extract stimulates interleukin-8 production in human airway epithelium and is attenuated by superoxide dismutase in vitro. Am. J. Rhinol. Allergy 2009, 23, e1–e4. [Google Scholar] [CrossRef]
- Doyon, W.M.; Dong, Y.; Ostroumov, A.; Thomas, A.M.; Zhang, T.A.; Dani, J.A. Nicotine decreases ethanol-induced dopamine signaling and increases self-administration via stress hormones. Neuron 2013, 79, 530–540. [Google Scholar] [CrossRef] [Green Version]
- Burnham, E.L.; McNally, A.; Gaydos, J.; Brown, L.A.S. The relationship between airway antioxidant levels, alcohol use disorders, and cigarette smoking. Alcohol Clin. Exp. Res. 2016, 40, 2147–2160. [Google Scholar] [CrossRef] [Green Version]
- Arcavi, L.; Benowitz, N.L. Cigarette smoking and infection. Arch. Intern. Med. 2004, 164, 2206–2216. [Google Scholar] [CrossRef]
- Pollard, M.S.; Tucker, J.S.; Green, H.D. Changes in adult alcohol use and consequences during the COVID-19 pandemic in the US. JAMA Netw. Open 2020, 3, e2022942. [Google Scholar] [CrossRef]
- Esposito, A.J.; Menon, A.A.; Ghosh, A.J.; Putman, R.K.; Fredenburgh, L.E.; El-Chemaly, S.Y.; Goldberg, H.J.; Baron, R.M.; Hunninghake, G.M.; Doyle, T.J. Increased odds of death for patients with interstitial lung disease and COVID-19: A case–control study. Am. J. Respir. Crit. Care Med. 2020, 202, 1710–1713. [Google Scholar] [CrossRef]
- Sheikh, D.; Tripathi, N.; Chandler, T.R.; Furmanek, S.; Bordon, J.; Ramirez, J.A.; Cavallazzi, R. Clinical outcomes in patients with COPD hospitalized with SARS-CoV-2 versus non-SARS-CoV-2 community-acquired pneumonia. Respir. Med. 2022, 191, 106714. [Google Scholar] [CrossRef]
- Calesnick, B.; Vernick, H. Antitussive activity of ethanol. Q. J. Stud. Alcohol 1971, 32, 434–441. [Google Scholar] [CrossRef]
- Sisson, J.H.; Wyatt, T.A.; Guidot, D.M.; Bagby, G.J.; Helander, A.; Tønnesen, H.; Spies, C.D. Bench to bedside: Mechanisms and consequences of alcohol-altered host defenses. Alcohol. Clin. Exp. Res. 2005, 29, 1090–1097. [Google Scholar] [CrossRef]
- Wu, D.; Cederbaum, A.I. Alcohol, oxidative stress, and free radical damage. Alcohol Res. Health 2003, 27, 277. [Google Scholar]
- Bailey, K.L.; Romberger, D.J.; Katafiasz, D.M.; Heires, A.J.; Sisson, J.H.; Wyatt, T.A.; Burnham, E.L. TLR2 and TLR4 Expression and Inflammatory Cytokines are Altered in the Airway Epithelium of Those with Alcohol Use Disorders. Alcohol Clin. Exp. Res. 2015, 39, 1691–1697. [Google Scholar] [CrossRef] [Green Version]
- Burnham, E.L.; Kovacs, E.J.; Davis, C.S. Pulmonary cytokine composition differs in the setting of alcohol use disorders and cigarette smoking. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L873–L882. [Google Scholar] [CrossRef] [Green Version]
- Gamble, L.; Mason, C.M.; Nelson, S. The effects of alcohol on immunity and bacterial infection in the lung. Med. Mal. Infect. 2006, 36, 72–77. [Google Scholar] [CrossRef]
- Wyatt, T.A.; Sisson, J.H.; Allen-Gipson, D.S.; McCaskill, M.L.; Boten, J.A.; DeVasure, J.M.; Bailey, K.L.; Poole, J.A. Co-exposure to cigarette smoke and alcohol decreases airway epithelial cell cilia beating in a protein kinase Cepsilon-dependent manner. Am. J. Pathol. 2012, 181, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Changeux, J.P.; Amoura, Z.; Rey, F.A.; Miyara, M. A nicotinic hypothesis for COVID-19 with preventive and therapeutic implications. C. R. Biol. 2020, 343, 33–39. [Google Scholar] [CrossRef]
- Tomchaney, M.; Contoli, M.; Mayo, J.; Baraldo, S.; Shuaizhi, L.; Cabel, C.; Bull, D.; Lick, S.; Malo, J.; Knoper, S. Paradoxical effects of cigarette smoke and COPD on SARS-CoV2 infection and disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Farsalinos, K.; Barbouni, A.; Poulas, K.; Polosa, R.; Caponnetto, P.; Niaura, R. Current smoking, former smoking, and adverse outcome among hospitalized COVID-19 patients: A systematic review and meta-analysis. Ther. Adv. Chronic Dis. 2020, 11, 2040622320935765. [Google Scholar] [CrossRef] [PubMed]
- Karanasos, A.; Aznaouridis, K.; Latsios, G.; Synetos, A.; Plitaria, S.; Tousoulis, D.; Toutouzas, K. Impact of Smoking Status on Disease Severity and Mortality of Hospitalized Patients With COVID-19 Infection: A Systematic Review and Meta-analysis. Nicotine Tob. Res. 2020, 22, 1657–1659. [Google Scholar] [CrossRef]
- Li, G.; He, X.; Zhang, L.; Ran, Q.; Wang, J.; Xiong, A.; Wu, D.; Chen, F.; Sun, J.; Chang, C. Assessing ACE2 expression patterns in lung tissues in the pathogenesis of COVID-19. J. Autoimmun. 2020, 112, 102463. [Google Scholar] [CrossRef]
- Ghosh, A.; Girish, V.; Yuan, M.L.; Coakley, R.D.; Wrennall, J.A.; Alexis, N.E.; Sausville, E.L.; Vasudevan, A.; Chait, A.R.; Sheltzer, J.M.; et al. Combustible and Electronic Cigarette Exposures Increase ACE2 Activity and SARS-CoV-2 Spike Binding. Am. J. Respir. Crit. Care Med. 2022, 205, 129–133. [Google Scholar] [CrossRef]
- Yeung, M.L.; Teng, J.L.L.; Jia, L.; Zhang, C.; Huang, C.; Cai, J.P.; Zhou, R.; Chan, K.H.; Zhao, H.; Zhu, L.; et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell 2021, 184, 2212–2228.E12. [Google Scholar] [CrossRef]
- Johansen, M.D.; Mahbub, R.M.; Idrees, S.; Nguyen, D.H.; Miemczyk, S.; Pathinayake, P.; Nichol, K.; Hansbro, N.G.; Gearing, L.J.; Hertzog, P.J.; et al. Increased SARS-CoV-2 Infection, Protease, and Inflammatory Responses in Chronic Obstructive Pulmonary Disease Primary Bronchial Epithelial Cells Defined with Single-Cell RNA Sequencing. Am. J. Respir. Crit. Care Med. 2022, 206, 712–729. [Google Scholar] [CrossRef]
- Ahn, J.H.; Kim, J.; Hong, S.P.; Choi, S.Y.; Yang, M.J.; Ju, Y.S.; Kim, Y.T.; Kim, H.M.; Rahman, M.D.T.; Chung, M.K.; et al. Nasal ciliated cells are primary targets for SARS-CoV-2 replication in the early stage of COVID-19. J. Clin. Invest. 2021, 131, e148517. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Nakayama, T.; Wu, C.T.; Goltsev, Y.; Jiang, S.; Gall, P.A.; Liao, C.K.; Shih, L.C.; Schurch, C.M.; McIlwain, D.R.; et al. ACE2 localizes to the respiratory cilia and is not increased by ACE inhibitors or ARBs. Nat. Commun. 2020, 11, 5453. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muralidharan, A.; Bauer, C.D.; Katafiasz, D.M.; Strah, H.M.; Siddique, A.; Reid, S.P.; Bailey, K.L.; Wyatt, T.A. Synergistic Detrimental Effects of Cigarette Smoke, Alcohol, and SARS-CoV-2 in COPD Bronchial Epithelial Cells. Pathogens 2023, 12, 498. https://doi.org/10.3390/pathogens12030498
Muralidharan A, Bauer CD, Katafiasz DM, Strah HM, Siddique A, Reid SP, Bailey KL, Wyatt TA. Synergistic Detrimental Effects of Cigarette Smoke, Alcohol, and SARS-CoV-2 in COPD Bronchial Epithelial Cells. Pathogens. 2023; 12(3):498. https://doi.org/10.3390/pathogens12030498
Chicago/Turabian StyleMuralidharan, Abenaya, Christopher D. Bauer, Dawn M. Katafiasz, Heather M. Strah, Aleem Siddique, St Patrick Reid, Kristina L. Bailey, and Todd A. Wyatt. 2023. "Synergistic Detrimental Effects of Cigarette Smoke, Alcohol, and SARS-CoV-2 in COPD Bronchial Epithelial Cells" Pathogens 12, no. 3: 498. https://doi.org/10.3390/pathogens12030498