

Molecular Characterization of the First African Swine Fever Virus Genotype II Strains Identified from Mainland Italy, 2022
 , ,
, ,  , , and
, , and 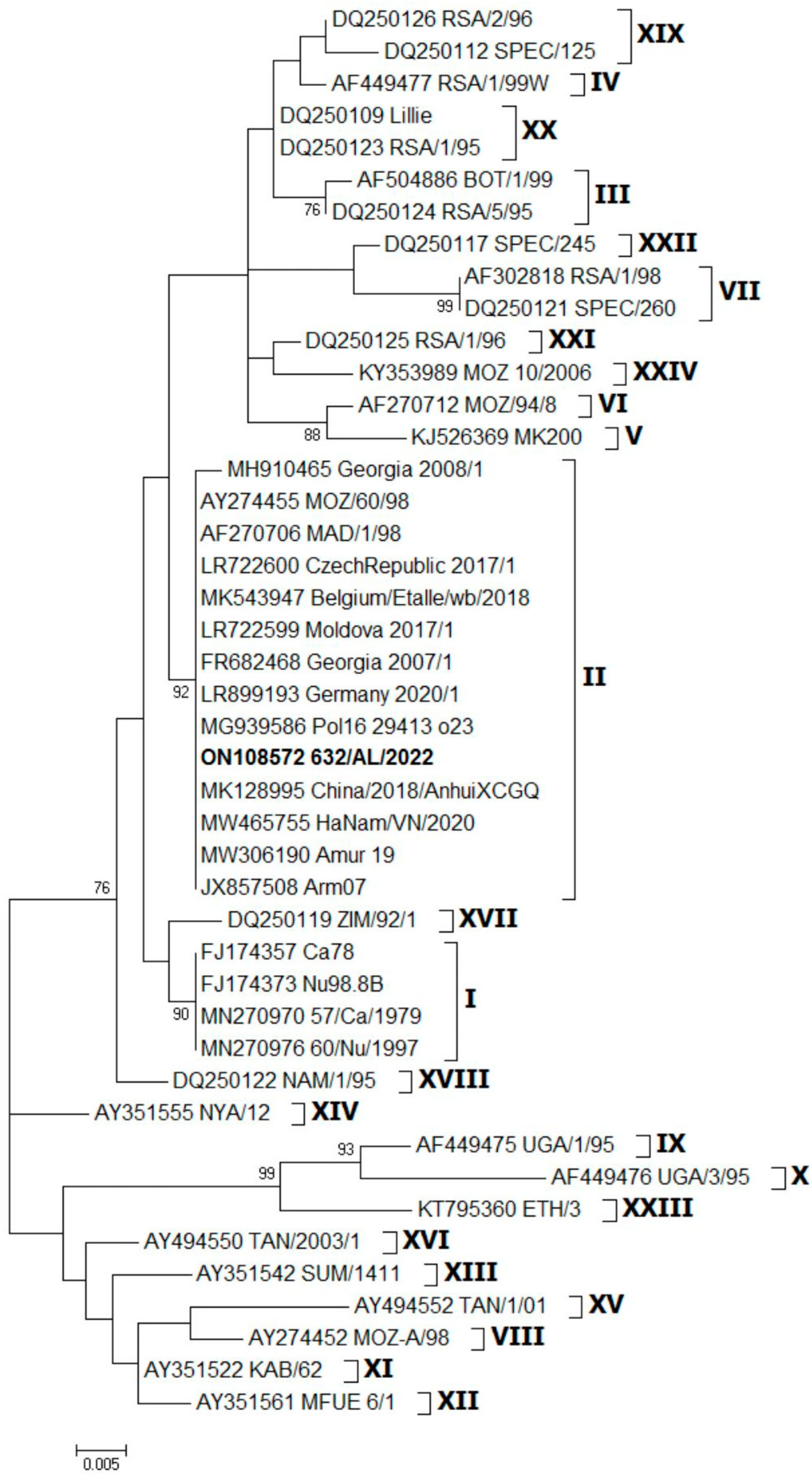{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Preparation
2.2. Partial Sequencing (Sanger)
2.3. Shotgun Metagenomic Analysis
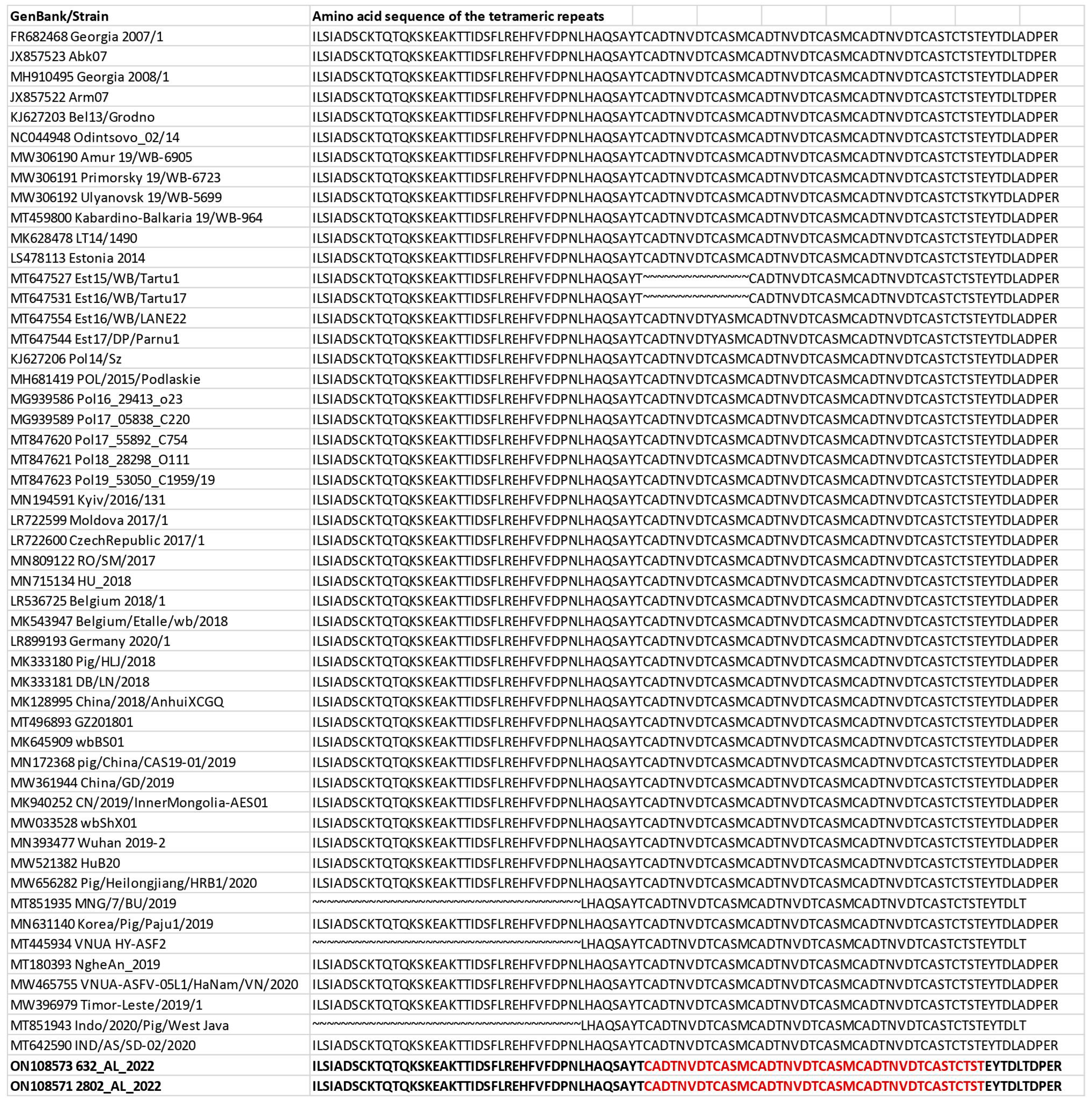
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Covadonga, A.; Borca, M.; Dixon, L.; Revilla, Y.; Rodriguez, F.; Escribano, J.M.; ICTV Consortium. ICTV Virus Taxonomy Profile. Asfarviridae. J. Gen. Virol. 2018, 99, 613–614. [Google Scholar] [CrossRef]
- Montgomery, R.E. On a form of swine fever occurring in british east-Africa (Kenya Colony). J. Comp. Pathol. Ther. 1921, 34, 159–191. [Google Scholar] [CrossRef] [Green Version]
- Danzetta, M.L.; Marenzoni, M.L.; Iannetti, S.; Tizzani, P.; Calistri, P.; Feliziani, F. African Swine fewer: Lessons to learn from past eradication experiences. A systematic review. Front. Vet. Sci. 2020, 7, 296. [Google Scholar] [CrossRef]
- Beltrán-Alcrudo, D.; Lubroth, J.; Depner, K.; De La Rocque, S. African swine fever in the Caucasus. FAO Empres Watch. 2008, 1, 1–8. [Google Scholar] [CrossRef]
- Iglesias, I.; Rodríguez, A.; Feliziani, F.; Rolesu, S.; De la Torre, A. Spatio-temporal Analysis of African Swine Fever in Sardinia (2012–2014): Trends in Domestic Pigs and Wild Boar. Transbound. Emerg. Dis. 2017, 64, 656–662. [Google Scholar] [CrossRef]
- Zhou, X.; Li, N.; Luo, Y.; Liu, Y.; Miao, F.; Chen, T.; Zhang, S.; Cao, P.; Li, X.; Tian, K.; et al. Emergence of African Swine Fever in China, 2018. Transbound. Emerg. Dis. 2018, 65, 1482–1484. [Google Scholar] [CrossRef] [Green Version]
- Kolbasov, D.; Titov, I.; Tsybanov, S.; Gogin, A.; Malogolovkin, A. African Swine Fever Virus, Siberia, Russia. Emerg. Infect. Dis. 2017, 24, 796–798. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations (FAO). ASF Situation in Asia & Pacific Update. 12 May 2022, 08:30 hours; Rome. Available online: https://www.fao.org/home/en (accessed on 12 May 2022).
- Rajukumar, K.; Senthilkumar, D.; Venkatesh, G.; Singh, F.; Patil, V.P.; Kombiah, S.; Tosh, C.; Dubey, C.K.; Sen, A.; Barman, N.N.; et al. Genetic characterization of African swine fever virus from domestic pigs in India. Transbound. Emerg. Dis. 2021, 68, 2687–2692. [Google Scholar] [CrossRef]
- Sauter-Louis, C.; Forth, J.H.; Probst, C.; Staubach, C.; Hlinak, A.; Rudovsky, A.; Holland, D.; Schlieben, P.; Goldner, M.; Schatz, J.; et al. Joining the club: First detection of African swine fever in wild boar in Germany. Transbound. Emerg. Dis. 2021, 68, 1744–1752. [Google Scholar] [CrossRef]
- Gonzales, W.; Moreno, C.; Duran, U.; Henao, N.; Bencosme, M.; Lora, P.; Reyes, R.; Nunez, R.; De Gracia, A.; Perez, A.M. African swine fever in the Dominican Republic. Transbound. Emerg. Dis. 2021, 68, 3018–3019. [Google Scholar] [CrossRef]
- Cima, G. African Swine Fever Confirmed in Haiti. JAVMA News, November 2001. Available online: https://www.avma.org/javma-news/2021-11-01/african-swine-fever-confirmed-haiti (accessed on 12 May 2022).
- Iscaro, C.; Dondo, A.; Ruocco, L.; Masoero, L.; Giammarioli, M.; Zoppi, S.; Guberti, V.; Feliziani, F. January 2022: Index case of new African Swine Fever incursion in mainland Italy. Transbound. Emerg. Dis. 2022, 69, 1707–1711. [Google Scholar] [CrossRef]
- Dixon, L.K.; Chapman, D.A.G.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef]
- Cackett, G.; Matelska, D.; Sýkora, M.; Portugal, R.; Malecki, M.; Bähler, J.; Dixon, L.; Werner, F. The African swine fever virus transcriptome. J. Virol. 2020, 94, e00119-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumption, K.J.; Hutchings, G.H.; Wilkinson, P.J.; Dixon, L.K. Variable regions on the genome of Malawi isolates of African swine fever virus. J. Gen. Virol. 1990, 71 Pt 10, 2331–2340. [Google Scholar] [CrossRef]
- Bastos, A.D.S.; Penrith, M.-L.; Macome, F.; Pinto, F.; Thomson, G.R. Co-circulation of two genetically distinct viruses in an outbreak of African swine fever in Mozambique: No evidence for individual co-infection. Vet. Microbiol. 2004, 103, 169–182. [Google Scholar] [CrossRef]
- Gallardo, C.; Okoth, E.; Pelayo, V.; Anchuelo, R.; Martín, E.; Simón, A.; Llorente, A.; Nieto, R.; Soler, A.; Martín, R.; et al. African swine fever viruses with two different genotypes, both of which occur in domestic pigs, are associated with ticks and adult warthogs, respectively, at a single geographical site. J. Gen. Virol. 2011, 92 Pt 2, 432–444. [Google Scholar] [CrossRef]
- Gallardo, C.; Mwaengo, D.M.; Macharia, J.M.; Arias, M.; Taracha, E.A.; Soler, A.; Okoth, E.; Martín, E.; Kasiti, J.; Bishop, R.P. Enhanced discrimination of African swine fever virus isolates through nucleotide sequencing of the p54, p72, and pB602L (CVR) genes. Virus Genes 2009, 38, 85–95. [Google Scholar] [CrossRef]
- Giammarioli, M.; Gallardo, C.; Oggiano, A.; Iscaro, C.; Nieto, R.; Pellegrini, C.; Dei Giudici, S.; Arias, M.; De Mia, G.M. Genetic characterisation of African swine fever viruses from recent and historical outbreaks in Sardinia (1978–2009). Virus Genes 2011, 42, 377–387. [Google Scholar] [CrossRef]
- Lubisi, B.A.; Duarte, A.; Bastos, S.; Dwarka, R.M.; Vosloo, W. Intra-genotypic resolution of African swine fever viruses from an East African domestic pig cycle: A combined p72-CVR approach. Virus Genes 2007, 35, 729–735. [Google Scholar] [CrossRef] [Green Version]
- Nix, R.J.; Gallardo, C.; Hutchings, G.; Blanco, E.; Dixon, K.L. Molecular epidemiology of African swine fever virus studied by analysis of four variable genome regions. Arch. Virol. 2006, 151, 2475–2494. [Google Scholar] [CrossRef]
- Portugal, R.; Coelho, J.; Hoper, D.; Little, N.S.; Smithson, C.; Upton, C.; Martins, C.; Leitao, A.; Gunther, M.K. Related strains of African swine fever virus with different virulence: Genome comparison and analysis. J. Gen. Virol. 2015, 96, 408–419. [Google Scholar] [CrossRef]
- Blasco, R.; Agüero, M.; Almendral, J.M.; Viñuela, E. Variable and constant regions in African swine fever virus DNA. Virology 1989, 168, 330–338. [Google Scholar] [CrossRef]
- Tabarés, E.; Olivares, I.; Santurde, G.; Garcia, M.J.; Martin, E.; Carnero, M.E. African swine fever virus DNA: Deletions and additions during adaptation to growth in monkey kidney cells. Arch. Virol. 1987, 97, 333–346. [Google Scholar] [CrossRef]
- Irusta, P.M.; Borca, M.V.; Kutish, G.F.; Lu, Z.; Caler, E.; Carrillo, C.; Rock, D.L. Amino acid tandem repeats within a late viral gene define the central variable region of African swine fever virus. Virology 1996, 220, 20–27. [Google Scholar] [CrossRef]
- Xiong, D.; Zhang, X.; Xiong, J.; Yu, J.; Wei, H. Rapid genome-wide sequence typing of African swine fever virus based on alleles. Virus Res. 2021, 297, 198357. [Google Scholar] [CrossRef]
- Bastos, A.D.; Penrith, M.L.; Cruciere, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; Thomson, G.R. Genotyping field strains of african swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef]
- Boshoff, C.I.; Bastos, A.D.; Gerber, L.J.; Vosloo, W. Genetic characterisation of african swine fever viruses from outbreaks in southern Africa (1973–1999). Vet. Microbiol. 2007, 121, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Achenbach, J.E.; Gallardo, C.; Nieto-Pelegrin, E.; Rivera-Arroyo, B.; Degefa-Negi, T.; Arias, M.; Jenberie, S.; Mulisa, D.D.; Gizaw, D.; Gelaye, E.; et al. Identification of a new genotype of african swine fever virus in domestic pigs from Ethiopia. Transbound. Emerg. Dis. 2016, 64, 1393–1404. [Google Scholar] [CrossRef]
- Rowlands, R.J.; Michaud, V.; Heath, L.; Hutchings, G.; Oura, C.; Vosloo, W.; Dwarka, R.; Onashvili, T.; Albina, E.; Dixon, L.K. African swine fever virus isolate, Georgia, 2007. Emerg. Infect. Dis. 2008, 14, 1870–1874. [Google Scholar] [CrossRef]
- Simulundu, E.; Chambaro, H.M.; Sinkala, Y.; Kajihara, M.; Ogawa, H.; Mori, A.; Ndebe, J.; Dautu, G.; Mataa, L.; Lubaba, C.H.; et al. Co-circulation of multiple genotypes of African swine fever viruses among domestic pigs in Zambia (2013–2015). Transbound. Emerg. Dis. 2018, 65, 114–122. [Google Scholar] [CrossRef]
- Sanna, G.; Dei Giudici, S.; Bacciu, D.; Angioi, P.P.; Giammarioli, M.; De Mia, G.M.; Oggiano, A. Improved strategy for molecular characterization of African swine fever viruses from Sardinia, based on analysis of p30, CD2V and I73R/I329L variable regions. Transbound. Emerg. Dis. 2017, 64, 1280–1286. [Google Scholar] [CrossRef]
- Onzere, C.K.; Bastos, A.D.; Okoth, E.A.; Lichoti, J.K.; Bochere, E.N.; Owido, M.G.; Ndambuki, G.; Bronsvoort, M.; Bishop, R.P. Multi-locus sequence typing of African swine fever viruses from endemic regions of Kenya and Eastern Uganda (2011–2013) reveals rapid B602L central variable region evolution. Virus Genes 2018, 54, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Mazur-Panasiuk, N.; Woźniakowski, G. The unique genetic variation within the O174L gene of Polish strains of African swine fever virus facilitates tracking virus origin. Adv. Virol. 2019, 164, 1667–1672. [Google Scholar] [CrossRef] [Green Version]
- Farlow, J.; Donduashvili, M.; Kokhreidze, M.; Kotorashvil, A.; Vepkhvadze, N.G.; Kotaria, N.; Gulbani, A. Intra-epidemic genome variation in highly pathogenic African swine fever virus (ASFV) from the country of Georgia. Virol. J. 2018, 15, 190. [Google Scholar] [CrossRef] [Green Version]
- Granberg, F.; Torresi, C.; Oggiano, A.; Malmberg, M.; Iscaro, C.; De Mia, G.M.; Belák, S. Complete genome sequence of an African swine fever virus isolate from Sardinia, Italy. Genome Announc. 2016, 4, e01220-16. [Google Scholar] [CrossRef] [Green Version]
- Masembe, C.; Sreenu, V.B.; Da Silva Filipe, A.; Wilkie, G.S.; Ogweng, P.; Mayega, F.J.; Muwanika, V.B.; Biek, R.; Palmarini, M.; Davison, A.J. Genome sequences of five African swine fever virus genotype IX isolates from domestic pigs in Uganda. Microbiol. Resour. Announc. 2018, 7, E01018-18. [Google Scholar] [CrossRef] [Green Version]
- Olesen, A.S.; Lohse, L.; Dalgaard, M.D.; Woźniakowski, G.; Belsham, G.J.; Bøtner, A.; Rasmussen, T.B. Complete genome sequence of an African swine fever virus (ASFV POL/2015/Podlaskie) determined directly from pig erythrocyte-associated nucleic acid. J. Virol. Methods 2018, 261, 14–16. [Google Scholar] [CrossRef] [Green Version]
- Mazur-Panasiuk, N.; Woźniakowski, G.; Niemczuk, K. The first complete genomic sequences of African swine fever virus isolated in Poland. Sci. Rep. 2019, 9, 4556. [Google Scholar] [CrossRef] [Green Version]
- Torresi, C.; Fiori, M.; Bertolotti, L.; Floris, M.; Colitti, B.; Giammarioli, M.; Dei Giudici, S.; Oggiano, A.; Malmberg, M.; De Mia, G.M.; et al. The evolution of African swine fever virus in Sardinia (1978–2014) as revealed by whole-genome sequencing and comparative analysis. Transbound. Emerg. Dis. 2020, 67, 1971–1980. [Google Scholar] [CrossRef]
- Enjuanes, L.; Carrascosa, A.L.; Viñuela, E. Isolation and properties of the DNA of African swine fever (ASF) virus. J. Gen. Virol. 1976, 32, 479–492. [Google Scholar] [CrossRef]
- Burland, T.G. DNASTAR’s Lasergene sequence analysis software. Methods Mol. Biol. 2000, 132, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit, a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 96116. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. UGENE team, Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [Green Version]
- Mannelli, A.; Sotgia, S.; Patta, C.; Sarria, A.; Madrau, P.; Sanna, L.; Firinu, A.; Laddomada, A. Effect of husbandry methods on seropositivity to African swine fever virus in Sardinian swine herds. Prev. Vet. Med. 1997, 32, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.M.; Salas, M.L.; Viñuela, E. Genes homologous to ubiquitin-conjugating proteins and eukaryotic transcription factor SII in African swine fever virus. Virology 1992, 186, 40–52. [Google Scholar] [CrossRef]
- Gallardo, C.; Fernández-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernández-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic variation among African swine fever genotype II viruses, eastern and central Europe. Emerg. Infect. Dis. 2014, 20, 1544–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goller, K.V.; Malogolovkin, A.S.; Katorkin, S.; Kolbasov, D.; Titov, I.; Höper, D.; Beer, M.; Keil, G.M.; Portugal, R.; Blome, S. Tandem repeat insertion in African swine fever virus, Russia, 2012. Emerg. Infect. Dis. 2015, 21, 731–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mur, L.; Atzeni, M.; Martínez-López, B.; Feliziani, F.; Rolesu, S.; Sanchez-Vizcaino, J.M. Thirty-Five-Year Presence of African Swine Fever in Sardinia: History, Evolution and Risk Factors for Disease Maintenance. Transbound. Emerg. Dis. 2016, 63, e165–e177. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giammarioli, M.; Alessandro, D.; Cammà, C.; Masoero, L.; Torresi, C.; Marcacci, M.; Zoppi, S.; Curini, V.; Rinaldi, A.; Rossi, E.; et al. Molecular Characterization of the First African Swine Fever Virus Genotype II Strains Identified from Mainland Italy, 2022. Pathogens 2023, 12, 372. https://doi.org/10.3390/pathogens12030372
Giammarioli M, Alessandro D, Cammà C, Masoero L, Torresi C, Marcacci M, Zoppi S, Curini V, Rinaldi A, Rossi E, et al. Molecular Characterization of the First African Swine Fever Virus Genotype II Strains Identified from Mainland Italy, 2022. Pathogens. 2023; 12(3):372. https://doi.org/10.3390/pathogens12030372
Chicago/Turabian StyleGiammarioli, Monica, Dondo Alessandro, Cesare Cammà, Loretta Masoero, Claudia Torresi, Maurilia Marcacci, Simona Zoppi, Valentina Curini, Antonio Rinaldi, Elisabetta Rossi, and et al. 2023. "Molecular Characterization of the First African Swine Fever Virus Genotype II Strains Identified from Mainland Italy, 2022" Pathogens 12, no. 3: 372. https://doi.org/10.3390/pathogens12030372