
Discovery and Development Strategies for SARS-CoV-2 NSP3 Macrodomain Inhibitors
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Materials, Reagents, and Chemicals
2.2. Constructs
2.3. Protein Expression and Purification for Crystallisation
2.4. HTRF Assay
2.5. Crystallisation, Crystal Soaking, and Data Processing
2.6. Virtual Screening
3. Results
3.1. NSP3 Macrodomain Virtual Ligand Screen
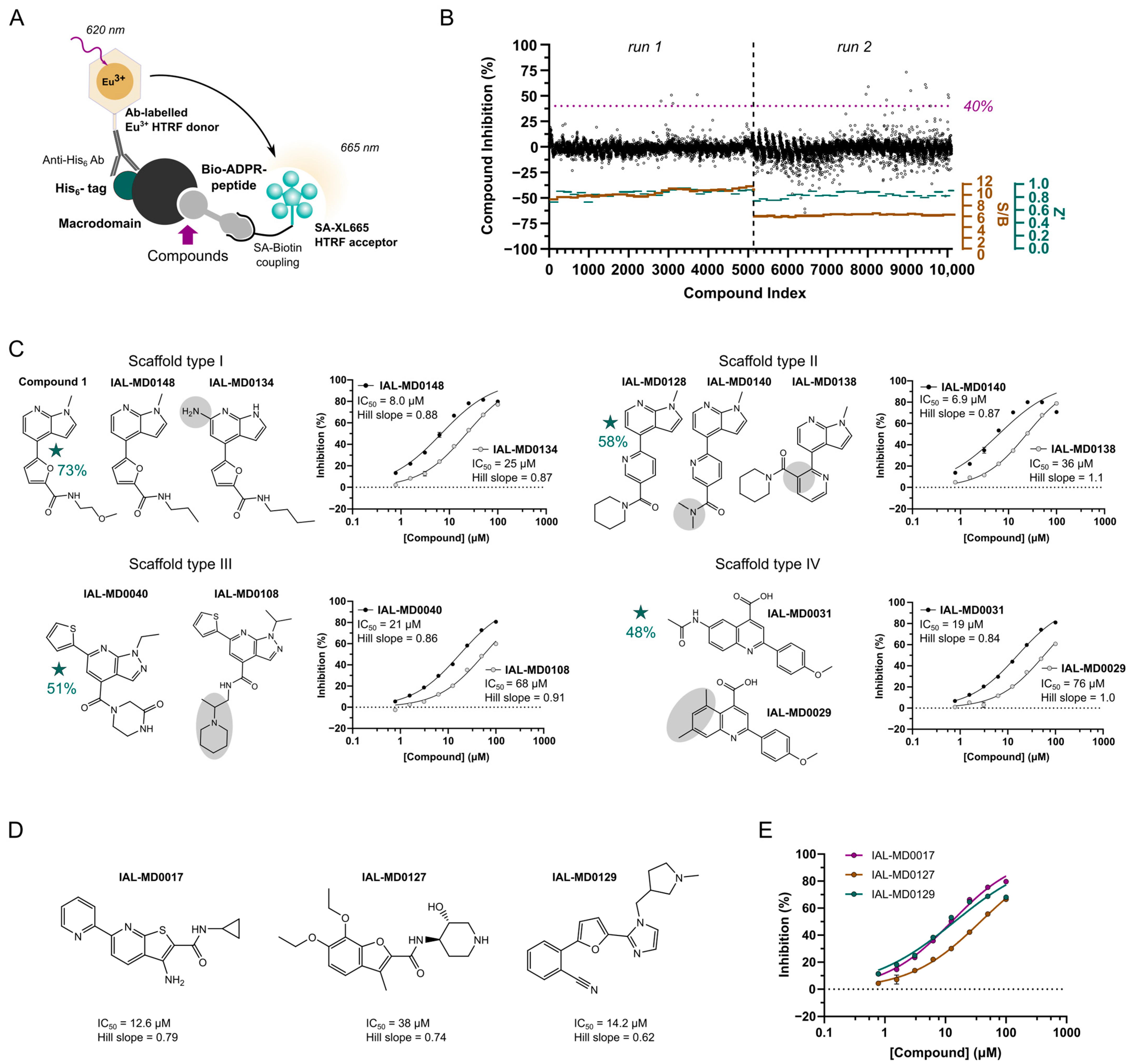
3.2. NSP3 Macrodomain Hit Discovery by MIDAS Compound Library Screen
3.3. NSP3 Macrodomain Inhibitors of Scaffold Type I
3.4. NSP3 Macrodomain Inhibitors of Scaffold Type II
3.5. NSP3 Macrodomain Inhibitors of Scaffold Type III
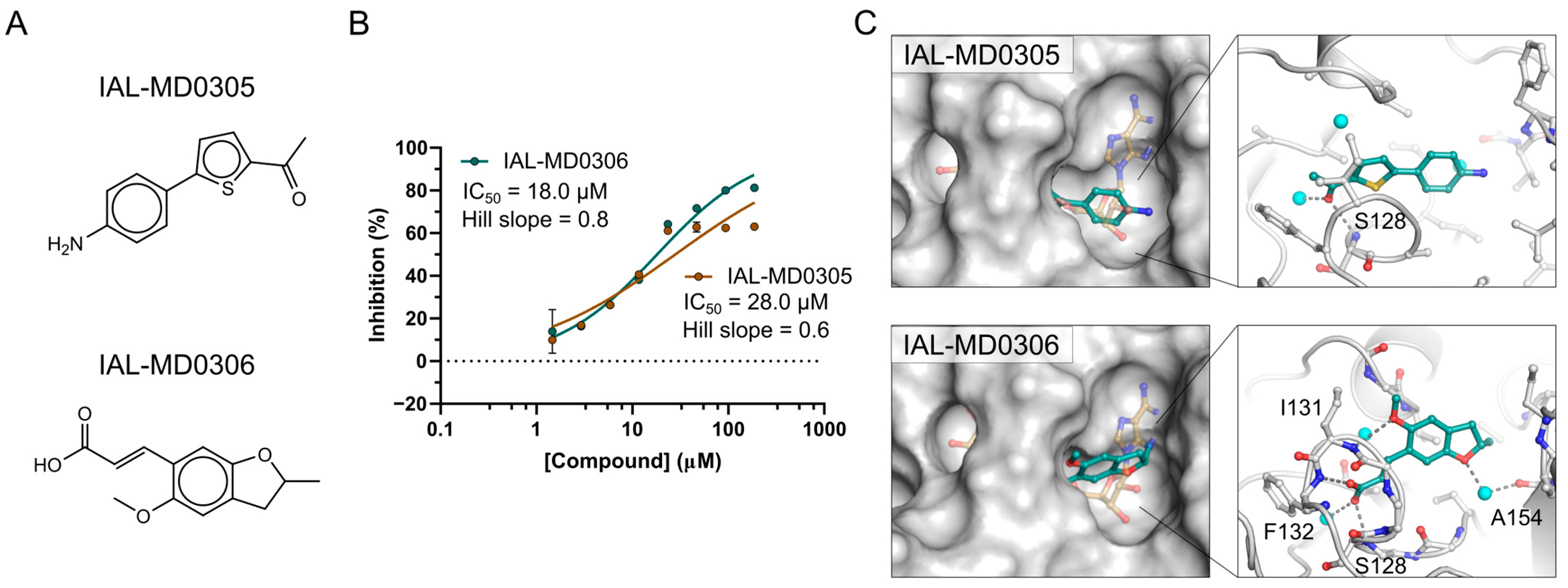
3.6. NSP3 Macrodomain Inhibitors of Scaffold Type IV and Singletons
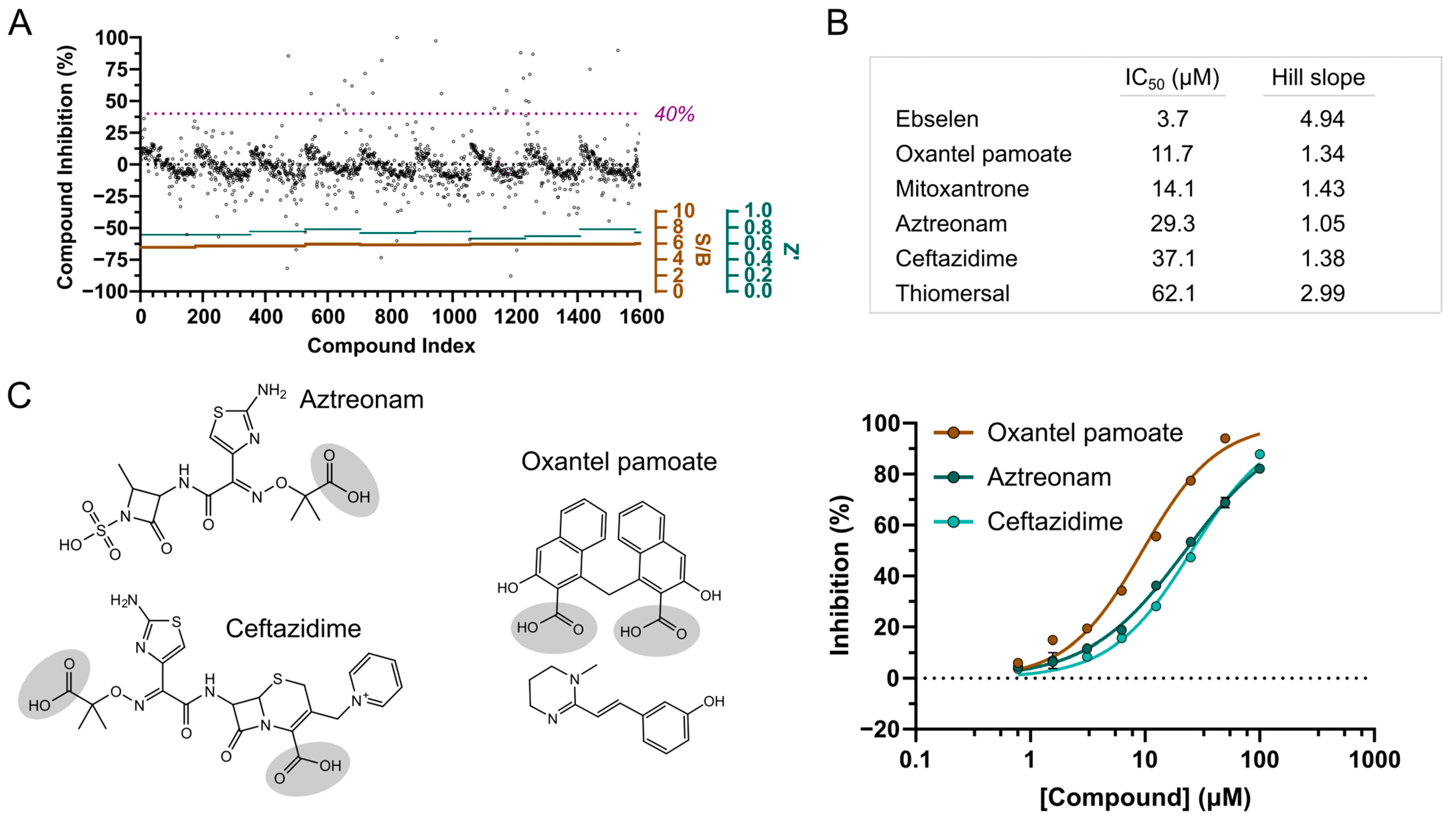
3.7. Screening of FDA-Approved Compounds Reveal Antibiotics as NSP3 Macrodomain Inhibitors
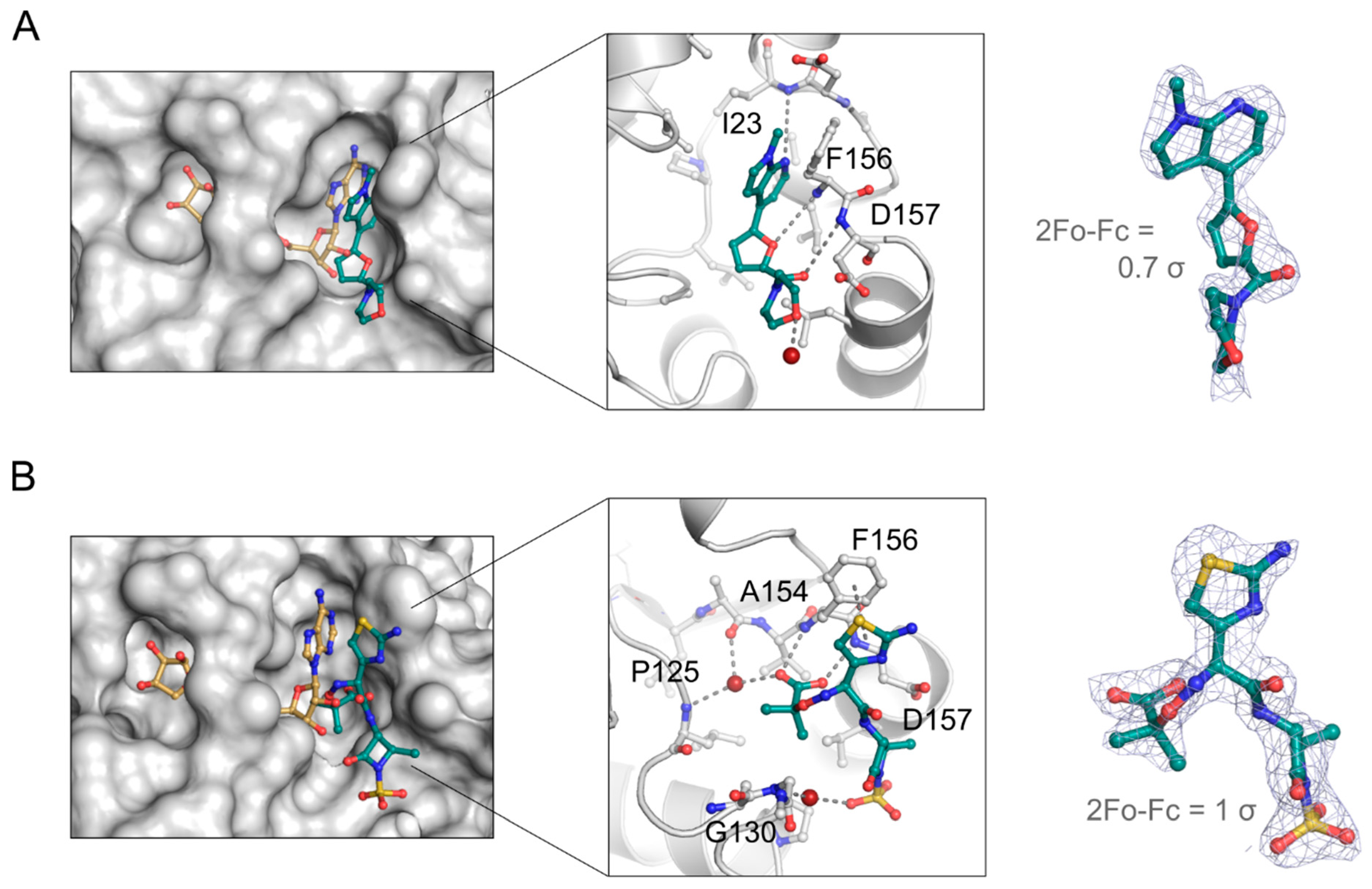
3.8. Aztreonam Targets the NSP3 Macrodomain Active Site Similar to MIDAS Hit Compound
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adam, D. True covid death toll could be more than double official count. Nature 2022, 605, 206. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Nicola, M.; Alsafi, Z.; Sohrabi, C.; Kerwan, A.; Al-jabir, A. The socio-economic implications of the coronavirus pandemic (COVID-19): A review. Int. J. Surg. J. 2020, 78, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Bai, C.; Zhong, Q.; Gao, G.F. Overview of SARS-CoV-2 genome-encoded proteins. Sci. China Life Sci. 2022, 65, 280–294. [Google Scholar] [CrossRef]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 540. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schafer, A.; Dinnon, K.H.; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef] [Green Version]
- Shyr, Z.A.; Gorshkov, K.; Chen, C.Z.; Zheng, W. Drug discovery strategies for sars-cov-2. J. Pharmacol. Exp. Ther. 2020, 375, 127–138. [Google Scholar] [CrossRef]
- Aiello, T.F.; García-Vidal, C.; Soriano, A. Antiviral drugs against SARS-CoV-2. Rev. Esp. Quimioter. 2022, 35, 10–15. [Google Scholar] [CrossRef]
- Cully, M. A tale of two antiviral targets—And the COVID-19 drugs that bind them. Nat. Rev. Drug Discov. 2022, 21, 3–5. [Google Scholar] [CrossRef]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res. 2020, 149, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, L.; Mikolčević, P.; Mikoč, A.; Ahel, I. ADP-ribosylation signalling and human disease. Open Biol. 2019, 9, 190041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rack, J.G.M.; Perina, D.; Ahel, I. Macrodomains: Structure, Function, Evolution, and Catalytic Activities. Annu. Rev. Biochem. 2016, 85, 431–454. [Google Scholar] [CrossRef] [PubMed]
- Schuller, M.; Ahel, I. Beyond protein modification: The rise of non-canonical ADP-ribosylation. Biochem. J. 2022, 479, 463–477. [Google Scholar] [CrossRef]
- Atasheva, S.; Frolova, E.I.; Frolov, I. Interferon-Stimulated Poly(ADP-Ribose) Polymerases Are Potent Inhibitors of Cellular Translation and Virus Replication. J. Virol. 2014, 88, 2116–2130. [Google Scholar] [CrossRef] [Green Version]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes Dev. 2020, 34, 341–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhao, H.; Liu, P.; Li, C.; Quanquin, N.; Ji, X.; Sun, N.; Du, P.; Qin, C.F.; Lu, N.; et al. PARP12 suppresses Zika virus infection through PARP-dependent degradation of NS1 and NS3 viral proteins. Sci. Signal. 2018, 11, eaas9332. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Mao, D.; Roswit, W.T.; Jin, X.; Patel, A.C.; Patel, D.A.; Agapov, E.; Wang, Z.; Tidwell, R.M.; Atkinson, J.J.; et al. PARP9-DTX3L ubiquitin ligase targets host histone H2BJ and viral 3C protease to enhance interferon signaling and control viral infection. Nat. Immunol. 2015, 16, 1215–1227. [Google Scholar] [CrossRef]
- Goenka, S.; Boothby, M. Selective potentiation of Stat-dependent gene expression by collaborator of Stat6 (CoaSt6), a transcriptional cofactor. PNAS 2006, 103, 4210–4215. [Google Scholar] [CrossRef] [Green Version]
- Iwata, H.; Goettsch, C.; Sharma, A.; Ricchiuto, P.; Goh, W.W.B.; Halu, A.; Yamada, I.; Yoshida, H.; Hara, T.; Wei, M.; et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat. Commun. 2016, 7, 12849. [Google Scholar] [CrossRef] [Green Version]
- Caprara, G.; Prosperini, E.; Piccolo, V.; Sigismondo, G.; Melacarne, A.; Cuomo, A.; Boothby, M.; Rescigno, M.; Bonaldi, T.; Natoli, G. PARP14 Controls the Nuclear Accumulation of a Subset of Type I IFN–Inducible Proteins. J. Immunol. 2018, 200, 2439–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, M.E.; Chen, Y.; Kuny, C.; Maejima, T.; Lease, R.; Ferraris, D.; Aikawa, M.; Sullivan, C.S.; Perlman, S.; Fehr, A.R. The coronavirus macrodomain is required to prevent PARP-mediated inhibition of virus replication and enhancement of IFN expression. PLoS Pathog. 2019, 15, e1007756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parthasarathy, S.; Fehr, A.R. PARP14: A key ADP-ribosylating protein in host–virus interactions? PLoS Pathog. 2022, 18, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Hoch, N.C. Host ADP-ribosylation and the SARS-CoV-2 macrodomain. Biochem. Soc. Trans. 2021, 49, 1711–1721. [Google Scholar] [CrossRef]
- Rack, J.M.G.; Zorzini, V.; Zhu, Z.; Schuller, M.; Ahel, D.; Ahel, I. Viral macrodomains: A structural and evolutionary assessment of the pharmacological potential. Open Biol. 2020, 10, 200237. [Google Scholar] [CrossRef]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.K.; Ahel, I.; Perlman, S. The conserved coronavirus macrodomain promotes virulence and suppresses the innate immune response during severe acute respiratory syndrome coronavirus infection. MBio 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Alhammad, Y.M.O.; Fehr, A.R. The viral macrodomain counters host antiviral ADP-ribosylation. Viruses 2020, 12, 384. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.K.L.; Griffin, D.E.; Bosch, J.; Fehr, A.R. The Conserved Macrodomain Is a Potential Therapeutic Target for Coronaviruses and Alphaviruses. Pathogens 2022, 11, 94. [Google Scholar] [CrossRef]
- Dasovich, M.; Zhuo, J.; Goodman, J.A.; Thomas, A.; McPherson, R.L.; Jayabalan, A.K.; Busa, V.F.; Cheng, S.J.; Murphy, B.A.; Redinger, K.R.; et al. High-Throughput Activity Assay for Screening Inhibitors of the SARS-CoV-2 Mac1 Macrodomain. ACS Chem. Biol. 2022, 17, 17–23. [Google Scholar] [CrossRef]
- Gahbauer, S.; Correyb, G.J.; Schuller, M.; Ferlad, M.P.; Dorukf, Y.U.; Rachmana, M.; Wug, T.; Diolaitif, M.; Wangh, S.; Neitzi, R.J.; et al. Iterative computational design and crystallographic screening identifies potent inhibitors targeting the Nsp3 macrodomain of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2023, 120, e2212931120. [Google Scholar] [CrossRef]
- Moiani, D.; Link, T.M.; Brosey, C.A.; Katsonis, P.; Lichtarge, O.; Kim, Y.; Joachimiak, A.; Ma, Z.; Kim, I.K.; Ahmed, Z.; et al. An Efficient Chemical Screening Method for Structure-Based Inhibitors to Nucleic Acid Enzymes Targeting the DNA Repair-Replication Interface and SARS CoV-2; Elsevier Inc.: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Roy, A.; Alhammad, Y.M.; Mcdonald, P.; Johnson, D.K.; Zhuo, J.; Leung, A.K.L.; Fehr, A.R. Discovery of compounds that inhibit SARS-CoV-2 Mac1-ADP-ribose binding by high-throughput screening. Antiviral Res. 2022, 203, 105344. [Google Scholar] [CrossRef] [PubMed]
- Correy, G.J.; Kneller, D.W.; Phillips, G.; Pant, S.; Russi, S.; Cohen, A.E.; Meigs, G.; Holton, J.M.; Gahbauer, S.; Thompson, M.C.; et al. The mechanisms of catalysis and ligand binding for the SARS-CoV-2 NSP3 macrodomain from neutron and x-ray diffraction at room temperature. Sci. Adv. 2022, 8, eabo5083. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Der, M.S.; Olieric, V.; Sharpe, M.E.; Hernandez-olmos, V.; Proschak, E.; Merk, D.; Knapp, S.; Chaikuad, A. Structural Insights into Plasticity and Discovery of Remdesivir Metabolite GS-441524 Binding in SARS-CoV - 2 Macrodomain. ACS Med. Chem. Lett. 2021, 12, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Schuller, M.; Correy, G.J.; Gahbauer, S.; Fearon, D.; Wu, T.; Díaz, R.E.; Young, I.D.; Carvalho Martins, L.; Smith, D.H.; Schulze-Gahmen, U.; et al. Fragment binding to the Nsp3 macrodomain of SARS-CoV-2 identified through crystallographic screening and computational docking. Sci. Adv. 2021, 7, eabf8711. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhong, F.; Li, M.; Liu, Y.; Wang, L.; Liu, M.; Li, F.; Zhang, J.; Wu, J.; Shi, Y.; et al. Two Binding Sites of SARS-CoV-2 Macrodomain 3 Probed by Oxaprozin and Meclomen. J. Med. Chem. 2022, 65, 15227–15237. [Google Scholar] [CrossRef]
- Babar, Z.; Khan, M.; Zahra, M.; Anwar, M.; Noor, K.; Hashmi, H.F.; Suleman, M.; Waseem, M.; Shah, A.; Ali, S.; et al. Drug similarity and structure-based screening of medicinal compounds to target macrodomain-I from SARS-CoV-2 to rescue the host immune system: A molecular dynamics study. J. Biomol. Struct. Dyn. 2022, 40, 523–537. [Google Scholar] [CrossRef]
- Patel, D.C.; Hausman, K.R.; Arba, M.; Tran, A.; Lakernick, P.M.; Wu, C. Novel inhibitors to ADP ribose phosphatase of SARS-CoV-2 identified by structure-based high throughput virtual screening and molecular dynamics simulations. Comput. Biol. Med. 2022, 140, 105084. [Google Scholar] [CrossRef]
- Brosey, C.A.; Houl, J.H.; Katsonis, P.; Jones, D.E.; Ahmed, Z.; Tainer, J.A. Targeting SARS-CoV-2 Nsp3 macrodomain structure with insights from human poly(ADP-ribose) glycohydrolase (PARG) structures with inhibitors. Prog. Biophys. Mol. Biol. 2021, 163, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.; Walters, M.A. Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, G. xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010, 43, 186–190. [Google Scholar] [CrossRef]
- Storoni, L.C.; McCoy, A.J.; Read, R.J. Likelihood-enhanced fast rotation functions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. 2010, D66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Schuller, M.; Riedel, K.; Gibbs-Seymour, I.; Uth, K.; Sieg, C.; Gehring, A.P.; Ahel, I.; Bracher, F.; Kessler, B.M.; Elkins, J.M.; et al. Discovery of a Selective Allosteric Inhibitor Targeting Macrodomain 2 of Polyadenosine-Diphosphate-Ribose Polymerase 14. ACS Chem. Biol. 2017, 12, 2866–2874. [Google Scholar] [CrossRef] [PubMed]
- Frick, D.N.; Virdi, R.S.; Vuksanovic, N.; Dahal, N.; Silvaggi, N.R. Molecular Basis for ADP-Ribose Binding to the Mac1 Domain of SARS-CoV-2 nsp3. Biochemistry 2020, 59, 2608–2615. [Google Scholar] [CrossRef] [PubMed]
- Iketani, S.; Mohri, H.; Culbertson, B.; Hong, S.J.; Duan, Y.; Luck, M.I.; Annavajhala, M.K.; Guo, Y.; Sheng, Z.; Uhlemann, A.; et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 2022, 613, 558–564. [Google Scholar] [CrossRef]
- Stevens, L.J.; Pruijssers, A.J.; Lee, H.W.; Gordon, C.J.; Tchesnokov, E.P.; Gribble, J.; George, A.S.; Hughes, T.M.; Lu, X.; Li, J.; et al. Mutations in the SARS-CoV-2 RNA-dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022, 14, eabo0718. [Google Scholar] [CrossRef]
- Virdi, R.S.; Bavisotto, R.V.; Hopper, N.C.; Vuksanovic, N.; Melkonian, T.R.; Silvaggi, N.R.; Frick, D.N. Discovery of Drug-Like Ligands for the Mac1 Domain of SARS-CoV-2 Nsp3. SLAS Discov. 2020, 25, 1162–1170. [Google Scholar] [CrossRef]
- Sowa, S.T.; Galera-Prat, A.; Wazir, S.; Alanen, H.I.; Maksimainen, M.M.; Lehtiö, L. A molecular toolbox for ADP-ribosyl binding proteins. Cell Rep. Methods 2021, 1, 100121. [Google Scholar] [CrossRef] [PubMed]
- Haikarainen, T.; Maksimainen, M.M.; Obaji, E.; Lehtiö, L. Development of an Inhibitor Screening Assay for Mono-ADP-Ribosyl Hydrolyzing Macrodomains Using AlphaScreen Technology. SLAS Discov. 2018, 23, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, I.; Pervaiz, N.; Khan, A.; Saleem, S.; Shireen, H.; Wei, D.Q.; Labrie, V.; Bao, Y.; Abbasi, A.A. Evolutionary and structural analysis of SARS-CoV-2 specific evasion of host immunity. Genes Immun. 2020, 21, 409–419. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuller, M.; Zarganes-Tzitzikas, T.; Bennett, J.; De Cesco, S.; Fearon, D.; von Delft, F.; Fedorov, O.; Brennan, P.E.; Ahel, I. Discovery and Development Strategies for SARS-CoV-2 NSP3 Macrodomain Inhibitors. Pathogens 2023, 12, 324. https://doi.org/10.3390/pathogens12020324
Schuller M, Zarganes-Tzitzikas T, Bennett J, De Cesco S, Fearon D, von Delft F, Fedorov O, Brennan PE, Ahel I. Discovery and Development Strategies for SARS-CoV-2 NSP3 Macrodomain Inhibitors. Pathogens. 2023; 12(2):324. https://doi.org/10.3390/pathogens12020324
Chicago/Turabian StyleSchuller, Marion, Tryfon Zarganes-Tzitzikas, James Bennett, Stephane De Cesco, Daren Fearon, Frank von Delft, Oleg Fedorov, Paul E. Brennan, and Ivan Ahel. 2023. "Discovery and Development Strategies for SARS-CoV-2 NSP3 Macrodomain Inhibitors" Pathogens 12, no. 2: 324. https://doi.org/10.3390/pathogens12020324