
Many Ways to Communicate—Crosstalk between the HBV-Infected Cell and Its Environment

{kind=link}
{kind=link}
{kind=link}
Abstract
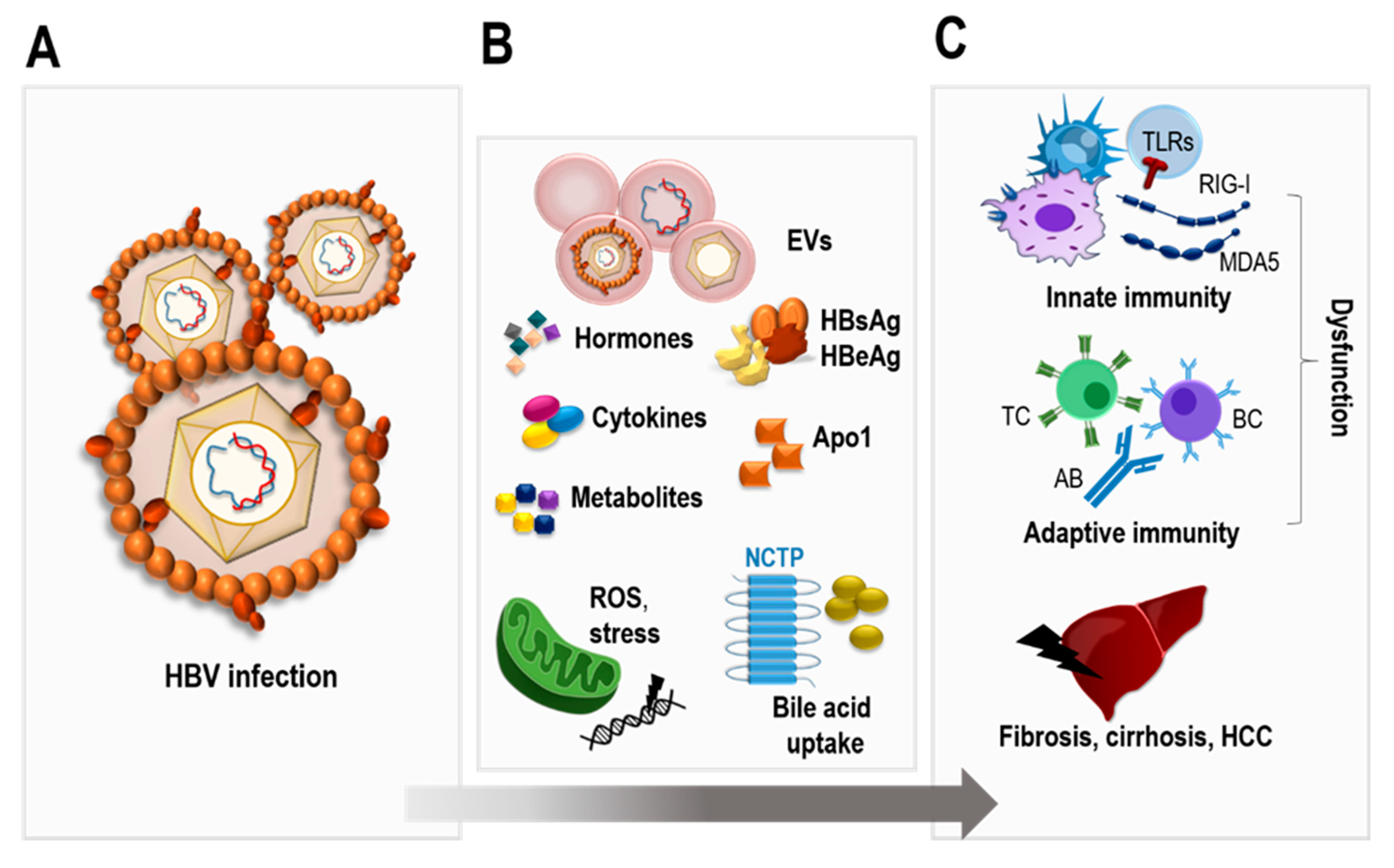
:1. Introduction
2. Molecular Biology of HBV
3. Interactions between HBV Infection, Metabolism, and Hormones
4. HBV and the Immune System
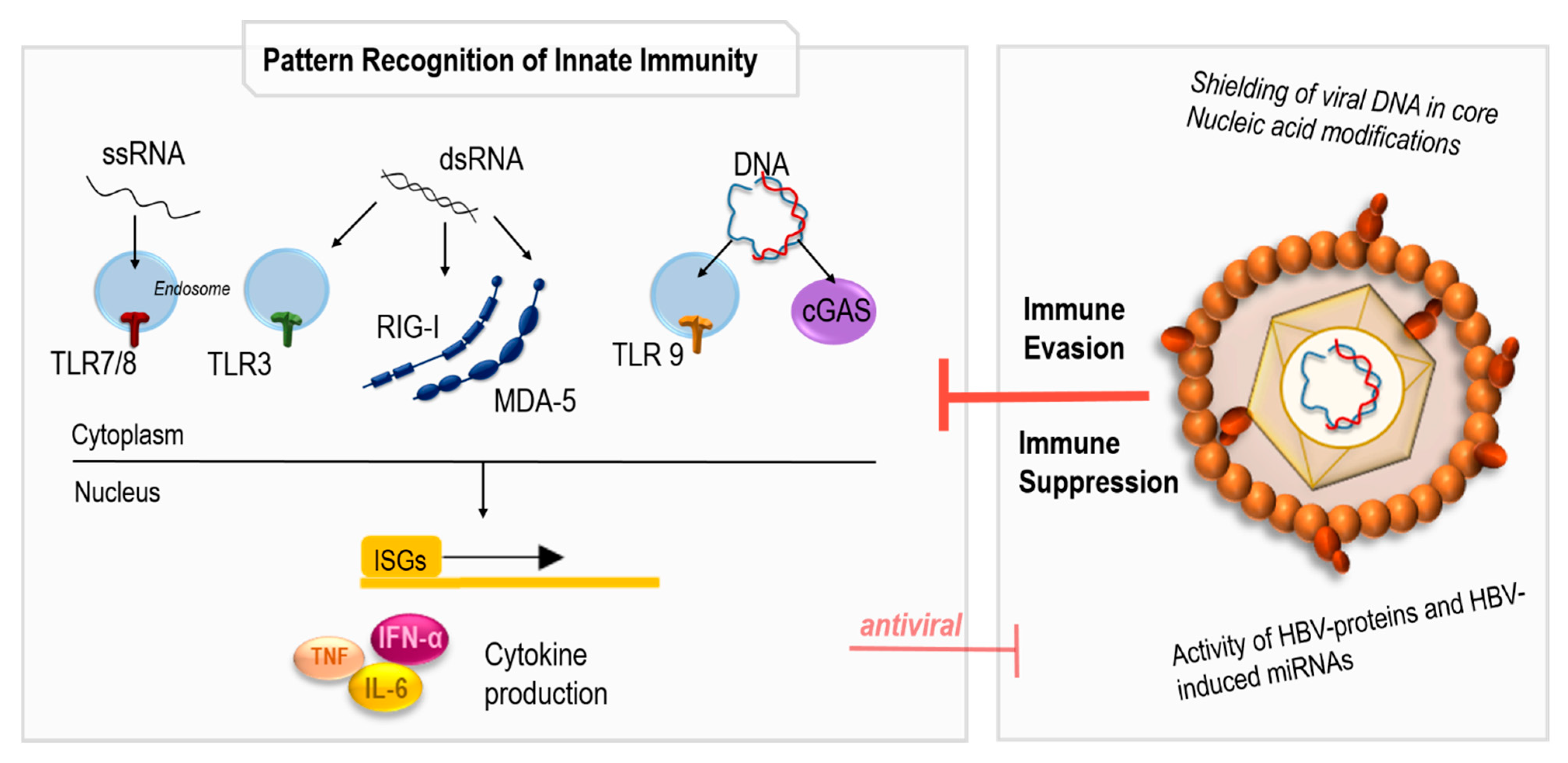
4.1. Sensing of HBV Infection by the Innate Immune System
4.2. Immune Response to HBV Infection
4.3. Role of Innate and Adaptive Immune Response and Related Cytokines on HBV-Related Carcinogenesis
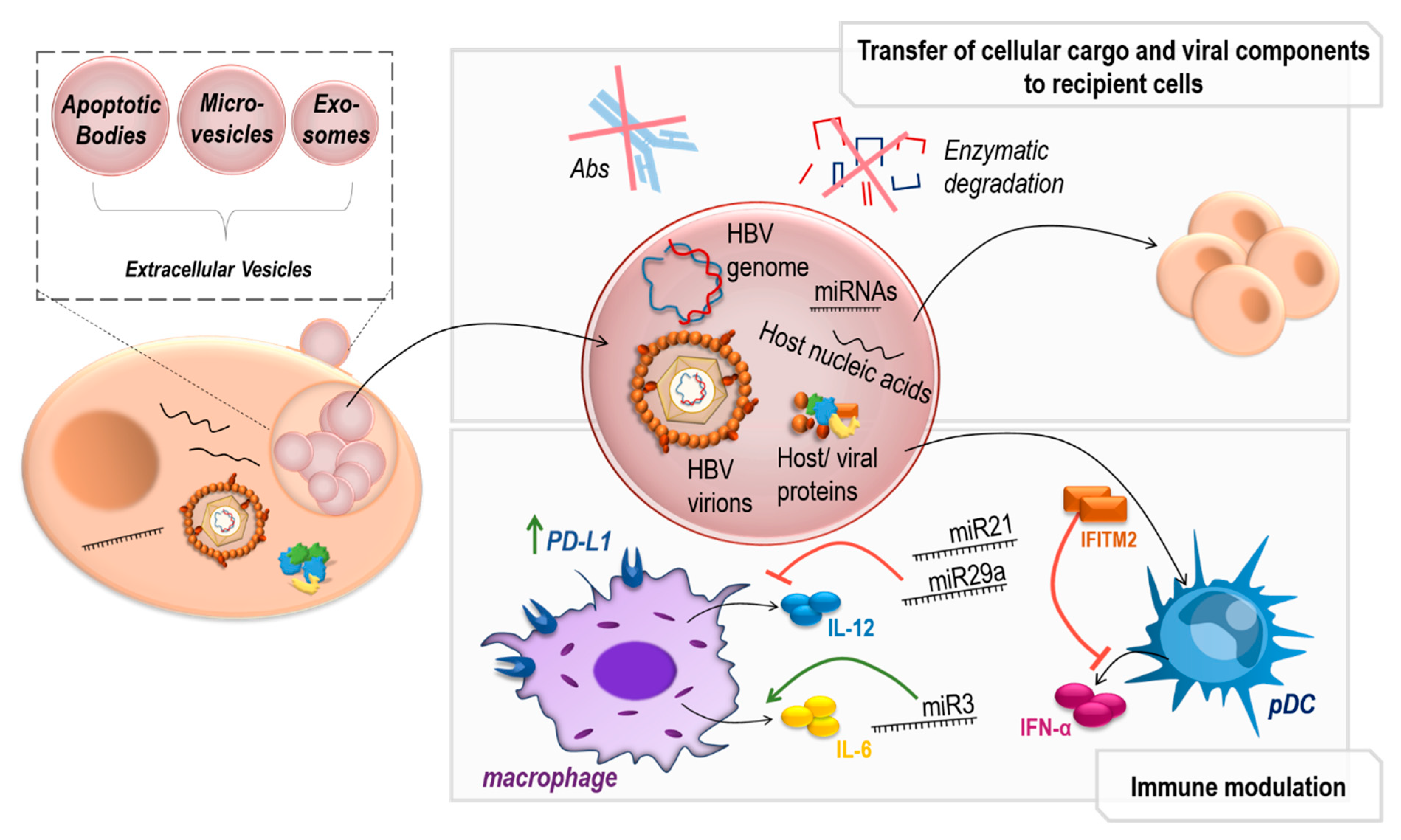
5. Extracellular Vesicles in HBV Infection
5.1. Extracellular Vesicles in Viral Infection
5.2. Immunomodulatory EVs in HBV Infection
5.3. Transfer of HBV Genomes in HBV-EVs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Global progress report on HIV, viral hepatitis and sexually transmitted infections. In Accountability for the Global Health Sector Strategies 2016–2021: Actions for Impact; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Bertoletti, A.; Ferrari, C. Innate and adaptive immune responses in chronic hepatitis B virus infections: Towards restoration of immune control of viral infection. Gut 2012, 61, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-C.; Sung, P.S.; Park, S.-H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Rubido, J.C.A.; Klundert, M.A.A.; Michel, M.L. Chronic Hepatitis B Treatment: Statu Quo and New Developments; Chapter 2 Hepatitis: A Global Health Concern; Semantical Scholar: Seattle, WA, USA, 2017; ISBN 978-93-87500-051. [Google Scholar]
- Noubiap, J.J.; Ndoula, S.T. Prevention of mother-to-child transmission of hepatitis B: Birth-dose vaccination is not enough. Lancet. Glob. Health 2022, 10, e455–e456. [Google Scholar] [CrossRef] [PubMed]
- Di Bisceglie, A.M. Hepatitis B and hepatocellular carcinoma. Hepatology 2009, 49 (Suppl. S5), S56–S60. [Google Scholar] [CrossRef] [Green Version]
- Chemin, I.; Zoulim, F. Hepatitis B virus induced hepatocellular carcinoma. Cancer Lett. 2009, 286, 52–59. [Google Scholar] [CrossRef]
- Kao, J.H. Hepatitis B Virus Genotypes and Hepatocellular Carcinoma in Taiwan. Intervirology 2003, 46, 400–407. [Google Scholar] [CrossRef]
- Golabi, P.; Fazel, S.; Otgonsuren, M.; Sayiner, M.; Locklear, C.T.; Younossi, Z.M. Mortality assessment of patients with hepatocellular carcinoma according to underlying disease and treatment modalities. Medicine 2017, 96, e5904. [Google Scholar] [CrossRef]
- Péneau, C.; Imbeaud, S.; La Bella, T.; Hirsch, T.Z.; Caruso, S.; Calderaro, J.; Paradis, T.; Blanc, J.-F.; Letouzé, E.; Nault, J.-C.; et al. Hepatitis B virus integrations promote local and distant oncogenic driver alterations in hepatocellular carcinoma. Gut 2022, 71, 616–626. [Google Scholar] [CrossRef]
- Zhao, K.; Liu, A.; Xia, Y. Insights into Hepatitis B Virus DNA Integration-55 Years after Virus Discovery. Innovation 2020, 1, 100034. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Decorsière, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.J.; Harris, J.M.; Marchi, E.; D’Arienzo, V.; Michler, T.; Wing, P.A.C.; Magri, A.; Ortega-Prieto, A.M.; van de Klundert, M.; Wettengel, J.; et al. Hypoxic gene expression in chronic hepatitis B virus infected patients is not observed in state-of-the-art in vitro and mouse infection models. Sci. Rep. 2020, 10, 14101. [Google Scholar] [CrossRef]
- Martín-Vílchez, S.; Sanz-Cameno, P.; Rodríguez-Muñoz, Y.; Majano, P.L.; Molina-Jiménez, F.; López-Cabrera, M.; Moreno-Otero, R.; Lara-Pezzi, E. The hepatitis B virus X protein induces paracrine activation of human hepatic stellate cells. Hepatology 2008, 47, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Tralhao, J.G.; Roudier, J.; Morosan, S.; Giannini, C.; Tu, H.; Goulenok, C.; Carnot, F.; Zavala, F.; Joulin, V.; Kremsdorf, D.; et al. Paracrine in vivo inhibitory effects of hepatitis B virus X protein (HBx) on liver cell proliferation: An alternative mechanism of HBx-related pathogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 6991–6996. [Google Scholar] [CrossRef] [Green Version]
- Quarleri, J. Core promoter: A critical region where the hepatitis B virus makes decisions. World J. Gastroenterol. 2014, 20, 425–435. [Google Scholar] [CrossRef]
- Kao, J.H.; Chen, P.J.; Lai, M.Y.; Chen, D.S. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology 2003, 124, 327–334. [Google Scholar] [CrossRef]
- Xie, X.; Luo, J.; Broering, R.; Zhu, D.; Zhou, W.; Lu, M.; Zheng, X.; Dittmer, U.; Yang, D.; Liu, J. HBeAg induces liver sinusoidal endothelial cell activation to promote intrahepatic CD8 T cell immunity and HBV clearance. Cell Mol. Immunol. 2021, 18, 2572–2574. [Google Scholar] [CrossRef]
- Erken, R.; Zaaijer, H.L.; Willemse, S.B.; Bakker, E.; Takkenberg, B.B.; Reesink, H.W.; Kootstra, N.A. Hepatitis B core related antigen in relation to intrahepatic and circulating viral markers, before and after combination therapy. Ann. Hepatol. 2021, 26, 100540. [Google Scholar] [CrossRef] [PubMed]
- Mak, L.Y.; Wong, D.K.; Cheung, K.S.; Seto, W.K.; Lai, C.L.; Yuen, M.F. Review article: Hepatitis B core-related antigen (HBcrAg): An emerging marker for chronic hepatitis B virus infection. Aliment. Pharmacol. Ther. 2018, 47, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Cai, D.; Liu, Y.; Guo, H. Detection of Hepatitis B Virus Particles Released from Cultured Cells by Particle Gel Assay. Methods Mol. Biol. 2017, 1540, 193–202. [Google Scholar] [PubMed]
- Lambert, C.; Döring, T.; Prange, R. Hepatitis B virus maturation is sensitive to functional inhibition of ESCRT-III, Vps4, and gamma 2-adaptin. J. Virol. 2007, 81, 9050–9060. [Google Scholar] [CrossRef] [Green Version]
- Prange, R. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med. Microbiol. Immunol. 2012, 201, 449–461. [Google Scholar] [CrossRef]
- Watanabe, T.; Sorensen, E.M.; Naito, A.; Schott, M.; Kim, S.; Ahlquist, P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci. USA 2007, 104, 10205–10210. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell. Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Tong, S.; Revill, P. Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 2016, 64 (Suppl. S1), S4–S16. [Google Scholar] [CrossRef] [Green Version]
- Patient, R.; Hourioux, C.; Sizaret, P.Y.; Trassard, S.; Sureau, C.; Roingeard, P. Hepatitis B virus subviral envelope particle morphogenesis and intracellular trafficking. J. Virol. 2007, 81, 3842–3851. [Google Scholar] [CrossRef] [Green Version]
- Zeyen, L.; Prange, R. Host Cell Rab GTPases in Hepatitis B Virus Infection. Front. Cell Dev. Biol. 2018, 6, 154. [Google Scholar] [CrossRef]
- Joo, E.J.; Cheong, H.S.; Kwon, M.J.; Sohn, W.; Kim, H.N.; Cho, Y.K. Relationship between gut microbiome diversity and hepatitis B viral load in patients with chronic hepatitis B. Gut Pathog. 2021, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Xia, P.; Zhou, X.; Li, X.; Guo, W.; Zhu, B.; Zheng, X.; Wang, B.; Yang, D.; Wang, J. Hepatitis B Virus Infection Alters Gut Microbiota Composition in Mice. Front. Cell Infect. Microbiol. 2019, 9, 377. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yi, X.; Yang, J.; Zhu, Z.; Wang, Y.; Liu, X.; Huang, X.; Wan, Y.; Fu, X.; Shu, W.; et al. Gut Microbiome Signatures in the Progression of Hepatitis B Virus-Induced Liver Disease. Front. Microbiol. 2022, 13, 916061. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, X.; Li, X.; Guo, W.; Zhu, Q.; Zhu, B.; Lu, Y.; Zheng, X.; Yang, D.; Wang, B. Fecal Microbiota Transplantation Alters the Outcome of Hepatitis B Virus Infection in Mice. Front. Cell Infect. Microbiol. 2022, 12, 844132. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.H.; Lim, M.K.; Oh, J.K.; Park, J.H.; Shin, A.; Sung, J.; Park, E.C. Combined effect of socioeconomic status, viral hepatitis, and lifestyles on hepatocelluar carcinoma risk in Korea. Br. J. Cancer 2010, 103, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.-F.; Cheng, Z.; Chisari, F.V.; Vu, T.H.; Hoffman, A.R.; Campbell, T.C. Repression of hepatitis B virus (HBV) transgene and HBV-induced liver injury by low protein diet. Oncogene 1997, 15, 2795–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.L.; Wu, Y.J.; Duan, Z.P.; Zhang, B.; Dong, P.L.; Ding, H.G. Resting energy expenditure and glucose, protein and fat oxidation in severe chronic virus hepatitis B patients. World J. Gastroenterol. 2008, 14, 4365–4369. [Google Scholar] [CrossRef]
- Shi, Y.-X.; Huang, C.-J.; Yang, Z.-G. Impact of hepatitis B virus infection on hepatic metabolic signaling pathway. World J. Gastroenterol. 2016, 22, 8161–8167. [Google Scholar] [CrossRef]
- Lamontagne, R.J.; Casciano, J.C.; Bouchard, M.J. A broad investigation of the HBV-mediated changes to primary hepatocyte physiology reveals HBV significantly alters metabolic pathways. Metabolism 2018, 83, 50–59. [Google Scholar] [CrossRef]
- Gao, Z.; Chen, J.; Zhou, Y.; Deng, P.; Sun, L.; Qi, J.; Zhang, P. A Novel Metabolism-Related Gene Signature for Predicting the Prognosis of HBV-Infected Hepatocellular Carcinoma. J. Oncol. 2022, 2022, 2391265. [Google Scholar] [CrossRef]
- Hossain, M.G.; Akter, S.; Ohsaki, E.; Ueda, K. Impact of the Interaction of Hepatitis B Virus with Mitochondria and Associated Proteins. Viruses 2020, 12, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- König, A.; Döring, B.; Mohr, C.; Geipel, A.; Geyer, J.; Glebe, D. Kinetics of the bile acid transporter and hepatitis B virus receptor Na+/taurocholate cotransporting polypeptide (NTCP) in hepatocytes. J. Hepatol. 2014, 61, 867–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oehler, N.; Volz, T.; Bhadra, O.D.; Kah, J.; Allweiss, L.; Giersch, K.; Bierwolf, J.; Riecken, K.; Pollok, J.M.; Lohse, A.W.; et al. Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology 2014, 60, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Hong, Y.; Che, Y.; He, T.; Wang, Y.; Zhang, S.; Wu, J.; Xu, W.; Hou, J.; Hao, H.; et al. Bile acid restrained T cell activation explains cholestasis aggravated hepatitis B virus infection. FASEB J. 2022, 36, e22468. [Google Scholar] [CrossRef]
- Zhang, J.; Ling, N.; Lei, Y.; Peng, M.; Hu, P.; Chen, M. Multifaceted Interaction Between Hepatitis B Virus Infection and Lipid Metabolism in Hepatocytes: A Potential Target of Antiviral Therapy for Chronic Hepatitis, B. Front. Microbiol. 2021, 12, 636897. [Google Scholar] [CrossRef]
- Hajjou, M.; Norel, R.; Carver, R.; Marion, P.; Cullen, J.; Rogler, L.E.; Rogler, C.E. cDNA microarray analysis of HBV transgenic mouse liver identifies genes in lipid biosynthetic and growth control pathways affected by HBV. J. Med. Virol. 2005, 77, 57–65. [Google Scholar] [CrossRef]
- Wang, Y.; Hao, J.; Liu, X.; Wang, H.; Zeng, X.; Yang, J.; Li, L.; Kuang, X.; Zhang, T. The mechanism of apoliprotein A1 down-regulated by Hepatitis B virus. Lipids Health Dis. 2016, 15, 64. [Google Scholar] [CrossRef] [Green Version]
- Baclig, M.O.; Reyes, K.G.; Liles, V.R.; Mapua, C.A.; Dimamay, M.P.S.; Gopez-Cervantes, J. Hepatic steatosis in chronic hepatitis B: A study of metabolic and genetic factors. Int. J. Mol. Epidemiol. Genet. 2018, 9, 13–19. [Google Scholar]
- Xiong, J.; Zhang, H.; Wang, Y.; Wang, A.; Bian, J.; Huang, H.; Zheng, Y.; Sang, X.; Xu, Y.; Lu, X.; et al. Hepatitis B virus infection and the risk of nonalcoholic fatty liver disease: A meta-analysis. Oncotarget 2017, 8, 107295–107302. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.; Goulder, P.; Matthews, P.C. Sexual Dimorphism in Chronic Hepatitis B Virus (HBV) Infection: Evidence to Inform Elimination Efforts. Wellcome Open Res. 2022, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Robinson, L.; Lee, N.L.; Welles, S.; Evans, A.A. No contribution of lifestyle and environmental exposures to gender discrepancy of liver disease severity in chronic hepatitis b infection: Observations from the Haimen City cohort. PLoS ONE 2017, 12, e0175482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.H.; Yeh, S.H.; Lin, W.H.; Yeh, K.H.; Yuan, Q.; Xia, N.S.; Chen, D.S.; Chen, P.J. Estrogen receptor α represses transcription of HBV genes via interaction with hepatocyte nuclear factor 4α. Gastroenterology 2012, 142, 989–998.e4. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.-S.; Au, K.-Y.; Fung, W.-C.; Wong, C.-Y.; Chan, A.C.-Y.; Lo, R.C.-L. Sex-specific analysis of microRNA profiles in HBV-associated cirrhosis by small RNA-sequencing. Hepatol. Commun. 2022, 12, 12,3473–3486. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef]
- Murphy, K.; Weaver, C.; Seidler, L. Janeway Immunologie; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Lind, N.A.; Rael, V.E.; Pestal, K.; Liu, B.; Barton, G.M. Regulation of the nucleic acid-sensing Toll-like receptors. Nat. Rev. Immunol. 2022, 22, 224–235. [Google Scholar] [CrossRef]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar]
- Bartok, E.; Hartmann, G. Immune Sensing Mechanisms that Discriminate Self from Altered Self and Foreign Nucleic Acids. Immunity 2020, 53, 54–77. [Google Scholar] [CrossRef]
- TThompson, M.G.; Sacco, M.T.; Horner, S.M. How RNA modifications regulate the antiviral response. Immunol. Rev. 2021, 304, 169–180. [Google Scholar] [CrossRef]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.F.; Chisari, F.V. Stealth and cunning: Hepatitis B and hepatitis C viruses. J. Virol. 2005, 79, 9369–9380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suslov, A.; Boldanova, T.; Wang, X.; Wieland, S.; Heim, M.H. Hepatitis B Virus Does Not Interfere With Innate Immune Responses in the Human Liver. Gastroenterology 2018, 154, 1778–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schöneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J.; et al. HBV Bypasses the Innate Immune Response and Does Not Protect HCV From Antiviral Activity of Interferon. Gastroenterology 2018, 154, 1791–1804.e22. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.W.; Imam, H.; Khan, M.; Siddiqui, A. N(6)-Methyladenosine modification of hepatitis B and C viral RNAs attenuates host innate immunity via RIG-I signaling. J. Biol. Chem. 2020, 295, 13123–13133. [Google Scholar] [CrossRef]
- Golsaz-Shirazi, F.; Shokri, F. Cross talk between hepatitis B virus and innate immunity of hepatocytes. Rev. Med. Virol. 2022, 32, e2256. [Google Scholar] [CrossRef]
- Zhao, H.J.; Hu, Y.F.; Han, Q.J.; Zhang, J. Innate and adaptive immune escape mechanisms of hepatitis B virus. World J. Gastroenterol. 2022, 28, 881–896. [Google Scholar] [CrossRef]
- Jung, S.; Altstetter, S.M.; Protzer, U. Innate immune recognition and modulation in hepatitis D virus infection. World J. Gastroenterol. 2020, 26, 2781–2791. [Google Scholar] [CrossRef]
- Wu, J.; Meng, Z.; Jiang, M.; Pei, R.; Trippler, M.; Broering, R.; Bucchi, A.; Sowa, J.P.; Dittmer, U.; Yang, D.; et al. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 2009, 49, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Aillot, L.; Bonnin, M.; Ait-Goughoulte, M.; Bendriss-Vermare, N.; Maadadi, S.; Dimier, L.; Subic, M.; Scholtes, C.; Najera, I.; Zoulim, F.; et al. Interaction between Toll-Like Receptor 9-CpG Oligodeoxynucleotides and Hepatitis B Virus Virions Leads to Entry Inhibition in Hepatocytes and Reduction of Alpha Interferon Production by Plasmacytoid Dendritic Cells. Antimicrob. Agents ChemoTher. 2018, 62, e01741-17. [Google Scholar] [CrossRef] [PubMed]
- Faure-Dupuy, S.; Lucifora, J.; Durantel, D. Interplay between the Hepatitis B Virus and Innate Immunity: From an Understanding to the Development of Therapeutic Concepts. Viruses 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandri, M.; Bertoletti, A.; Lütgehetmann, M. Innate immunity in hepatitis B and D virus infection: Consequences for viral persistence, inflammation, and T cell recognition. Semin. Immunopathol. 2021, 43, 535–548. [Google Scholar] [CrossRef]
- Burton, A.R.; Pallett, L.J.; McCoy, L.E.; Suveizdyte, K.; Amin, O.E.; Swadling, L.; Alberts, E.; Davidson, B.R.; Kennedy, P.T.; Gill, U.S.; et al. Circulating and intrahepatic antiviral B cells are defective in hepatitis B. J. Clin. Investig. 2018, 128, 4588–4603. [Google Scholar] [CrossRef] [Green Version]
- Michler, T.; Kosinska, A.D.; Festag, J.; Bunse, T.; Su, J.; Ringelhan, M.; Imhof, H.; Grimm, D.; Steiger, K.; Mogler, C.; et al. Knockdown of Virus Antigen Expression Increases Therapeutic Vaccine Efficacy in High-Titer Hepatitis B Virus Carrier Mice. Gastroenterology 2020, 158, 1762–1775.e9. [Google Scholar] [CrossRef]
- Stelma, F.; Willemse, S.B.; Erken, R.; de Niet, A.; Sinnige, M.J.; van Dort, K.; Zaaijer, H.L.; van Leeuwen, E.M.M.; Kootstra, N.A.; Reesink, H.W. Dynamics of the Immune Response in Acute Hepatitis B Infection. Open Forum Infect. Dis. 2017, 4, ofx231. [Google Scholar] [CrossRef] [Green Version]
- Rehermann, B.; Lau, D.; Hoofnagle, J.H.; Chisari, F.V. Cytotoxic T lymphocyte responsiveness after resolution of chronic hepatitis B virus infection. J. Clin. Investig. 1996, 97, 1655–1665. [Google Scholar] [CrossRef] [Green Version]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef]
- Stadler, D.; Kächele, M.; Jones, A.N.; Hess, J.; Urban, C.; Schneider, J.; Xia, Y.; Oswald, A.; Nebioglu, F.; Bester, R.; et al. Interferon-induced degradation of the persistent hepatitis B virus cccDNA form depends on ISG20. EMBO Rep. 2021, 22, e49568. [Google Scholar] [CrossRef]
- Liaw, Y.-F.; Chu, C.-M. Hepatitis B virus infection. Lancet 2009, 373, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Kakimi, K.; Wieland, S.; Guidotti, L.G.; Chisari, F.V. Interleukin-18 inhibits hepatitis B virus replication in the livers of transgenic mice. J. Virol. 2002, 76, 10702–10707. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Liu, J.; Wu, J.; Zhu, Y.; Li, G.; Wang, J.; Luo, M.; Deng, Q.; Zhang, J.; Xie, Y. IL-21-based therapies induce clearance of hepatitis B virus persistence in mouse models. Theranostics 2019, 9, 3798–3811. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Shen, Z.; Wu, J.; Song, Y.; Liu, N.; Deng, Q.; Xie, Y.; Liu, J. Interleukin-33 mediates both immune-related and non-immune-related inhibitory effects against hepatitis B virus. Antivir. Res. 2022, 206, 105404. [Google Scholar] [CrossRef]
- Wu, J.F.; Wu, T.C.; Chen, C.H.; Ni, Y.H.; Chen, H.L.; Hsu, H.Y.; Chang, M.H. Serum levels of interleukin-10 and interleukin-12 predict early, spontaneous hepatitis B virus e antigen seroconversion. Gastroenterology 2010, 138, 165–172.e1-3. [Google Scholar] [CrossRef]
- Rybicka, M.; Woziwodzka, A.; Sznarkowska, A.; Romanowski, T.; Stalke, P.; Dręczewski, M.; Verrier, E.R.; Baumert, T.F.; Bielawski, K.P. Genetic variation in IL-10 influences the progression of hepatitis B infection. Int. J. Infect. Dis. 2020, 96, 260–265. [Google Scholar] [CrossRef]
- Nieters, A.; Yuan, J.M.; Sun, C.L.; Zhang, Z.Q.; Stoehlmacher, J.; Govindarajan, S.; Yu, M.C. Effect of cytokine genotypes on the hepatitis B virus-hepatocellular carcinoma association. Cancer 2005, 103, 740–748. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Guilhot, S.; Chisari, F.V. Interleukin-2 and alpha/beta interferon down-regulate hepatitis B virus gene expression in vivo by tumor necrosis factor-dependent and -independent pathways. J. Virol. 1994, 68, 1265–1270. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.; Ma, J.; Jia, S.; Yang, L.; Wang, W.; Jin, Z. Interleukin-35 Suppresses Antiviral Immune Response in Chronic Hepatitis B Virus Infection. Front. Cell Infect. Microbiol. 2017, 7, 472. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Liu, Y.; Chen, Z.; Zheng, M. Involvement of Interleukin 6 in Hepatitis B Viral Infection. Cell Physiol. Biochem. 2015, 37, 677–686. [Google Scholar] [CrossRef]
- Hösel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Horiguchi, N.; Mori, M.; Gao, B. Cytokines and STATs in Liver Fibrosis. Front. Physiol. 2012, 3, 69. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Cheung, S.T. STAT3: An Emerging Therapeutic Target for Hepatocellular Carcinoma. Cancer 2019, 11, 1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.Y.; Yang, S.L.; Liang, H.F.; Li, C.H. HBx protein promotes oval cell proliferation by up-regulation of cyclin D1 via activation of the MEK/ERK and PI3K/Akt pathways. Int. J. Mol. Sci. 2014, 15, 3507–3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hösel, M.; Quasdorff, M.; Ringelhan, M.; Kashkar, H.; Debey-Pascher, S.; Sprinzl, M.F.; Bockmann, J.H.; Arzberger, S.; Webb, D.; von Olshausen, G.; et al. Hepatitis B Virus Activates Signal Transducer and Activator of Transcription 3 Supporting Hepatocyte Survival and Virus Replication. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 339–363. [Google Scholar] [CrossRef]
- Choudhari, S.R.; Khan, M.A.; Harris, G.; Picker, D.; Jacob, G.S.; Block, T.; Shailubhai, K. Deactivation of Akt and STAT3 signaling promotes apoptosis, inhibits proliferation, and enhances the sensitivity of hepatocellular carcinoma cells to an anticancer agent, Atiprimod. Mol. Cancer Ther. 2007, 6, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Xiang, W.Q.; Feng, W.F.; Ke, W.; Sun, Z.; Chen, Z.; Liu, W. Hepatitis B virus X protein stimulates IL-6 expression in hepatocytes via a MyD88-dependent pathway. J. Hepatol. 2011, 54, 26–33. [Google Scholar] [CrossRef]
- Zhang, L.J.; Wang, X.Z. Interleukin-10 and chronic liver disease. World J. Gastroenterol. 2006, 12, 1681–1685. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Díaz, D. Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [Green Version]
- Kouwaki, T.; Fukushima, Y.; Daito, T.; Sanada, T.; Yamamoto, N.; Mifsud, E.J.; Leong, C.R.; Tsukiyama-Kohara, K.; Kohara, M.; Matsumoto, M.; et al. Extracellular Vesicles Including Exosomes Regulate Innate Immune Responses to Hepatitis B Virus Infection. Front. Immunol. 2016, 7, 335. [Google Scholar] [CrossRef] [Green Version]
- Kouwaki, T.; Okamoto, M.; Tsukamoto, H.; Fukushima, Y.; Oshiumi, H. Extracellular Vesicles Deliver Host and Virus RNA and Regulate Innate Immune Response. Int. J. Mol. Sci. 2017, 18, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Porcellati, S.; Emiliani, C. The Role of Extracellular Vesicles in Viral Infection and Transmission. Vaccines 2019, 7, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raab-Traub, N.; Dittmer, D.P. Viral effects on the content and function of extracellular vesicles. Nat. Rev. Microbiol. 2017, 15, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolte-‘t Hoen, E.; Cremer, T.; Gallo, R.C.; Margolis, L.B. Extracellular vesicles and viruses: Are they close relatives? Proc. Natl. Acad. Sci. USA 2016, 113, 9155–9161. [Google Scholar] [CrossRef] [Green Version]
- van der Grein, S.G.; Defourny, K.A.Y.; Slot, E.F.J.; Nolte-’t Hoen, E.N.M. Intricate relationships between naked viruses and extracellular vesicles in the crosstalk between pathogen and host. Semin Immunopathol. 2018, 40, 491–504. [Google Scholar] [CrossRef] [Green Version]
- van der Grein, S.G.; Defourny, K.A.Y.; Rabouw, H.H.; Galiveti, C.R.; Langereis, M.A.; Wauben, M.H.M.; Arkesteijn, G.J.A.; van Kuppeveld, F.J.M.; Nolte-’t Hoen, E.N.M. Picornavirus infection induces temporal release of multiple extracellular vesicle subsets that differ in molecular composition and infectious potential. PLoS Pathog. 2019, 15, e1007594. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Li, Y.; McKnight, K.L.; Hensley, L.; Lanford, R.E.; Walker, C.M.; Lemon, S.M. Human pDCs preferentially sense enveloped hepatitis A virions. J. Clin. Investig. 2015, 125, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, S.; Takahashi, M.; Kobayashi, T.; Nishizawa, T.; Nishiyama, T.; Primadharsini, P.P.; Okamoto, H. Characterization of the Quasi-Enveloped Hepatitis E Virus Particles Released by the Cellular Exosomal Pathway. J. Virol. 2017, 91, e00822-17. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Wu, J.; Fang, D.; Qiu, Y.; Zou, X.; Jia, X.; Yin, Y.; Shen, L.; Mao, L. Exosomes cloak the virion to transmit Enterovirus 71 non-lytically. Virulence 2020, 11, 32–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Li, S.; Wu, S.; Chen, L. Exosomes Modulate the Viral Replication and Host Immune Responses in HBV Infection. Biomed. Res. Int. 2019, 2019, 2103943. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Du, L.; Lv, D.; Li, Y.; Zhang, Z.; Huang, X.; Tang, H. Emerging role and therapeutic application of exosome in hepatitis virus infection and associated diseases. J. Gastroenterol. 2021, 56, 336–349. [Google Scholar] [CrossRef]
- Kerviel, A.; Zhang, M.; Altan-Bonnet, N. A New Infectious Unit: Extracellular Vesicles Carrying Virus Populations. Annu. Rev. Cell. Dev. Biol. 2021, 37, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Décembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Grünvogel, O.; Colasanti, O.; Lee, J.Y.; Klöss, V.; Belouzard, S.; Reustle, A.; Esser-Nobis, K.; Hesebeck-Brinckmann, J.; Mutz, P.; Hoffmann, K.; et al. Secretion of Hepatitis C Virus Replication Intermediates Reduces Activation of Toll-Like Receptor 3 in Hepatocytes. Gastroenterology 2018, 154, 2237–2251.e16. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, K. Complete and Incomplete Hepatitis B Virus Particles: Formation, Function, and Application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.; Himmelsbach, K.; Ren, H.; Boller, K.; Hildt, E. Subviral Hepatitis B Virus Filaments, like Infectious Viral Particles, Are Released via Multivesicular Bodies. J. Virol. 2015, 90, 3330–3341. [Google Scholar] [CrossRef] [Green Version]
- Kakizaki, M.; Yamamoto, Y.; Otsuka, M.; Kitamura, K.; Ito, M.; Kawai, H.D.; Muramatsu, M.; Kagawa, T.; Kotani, A. Extracellular vesicles secreted by HBV-infected cells modulate HBV persistence in hydrodynamic HBV transfection mouse model. J. Biol. Chem. 2020, 295, 12449–12460. [Google Scholar] [CrossRef]
- Kakizaki, M.; Yamamoto, Y.; Yabuta, S.; Kurosaki, N.; Kagawa, T.; Kotani, A. The immunological function of extracellular vesicles in hepatitis B virus-infected hepatocytes. PLoS ONE 2018, 13, e0205886. [Google Scholar] [CrossRef]
- Huang, Z.Y.; Xu, P.; Li, J.H.; Zeng, C.H.; Song, H.F.; Chen, H.; Zhu, Y.B.; Song, Y.Y.; Lu, H.L.; Shen, C.P.; et al. Clinical Significance of Dynamics of Programmed Death Ligand-1 Expression on Circulating CD14(+) Monocytes and CD19(+) B Cells with the Progression of Hepatitis B Virus Infection. Viral Immunol. 2017, 30, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Du, L.; Lv, D.; Li, H.; Shang, J.; Lu, J.; Zhou, L.; Bai, L.; Tang, H. Exosomal Interferon-Induced Transmembrane Protein 2 Transmitted to Dendritic Cells Inhibits Interferon Alpha Pathway Activation and Blocks Anti-Hepatitis B Virus Efficacy of Exogenous Interferon Alpha. Hepatology 2019, 69, 2396–2413. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, N.R.; Chadha, R.; Kumar, S.; Choedon, T.; Reddy, V.S.; Kumar, V. The HBx gene of hepatitis B virus can influence hepatic microenvironment via exosomes by transferring its mRNA and protein. Virus Res. 2017, 240, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhao, X.; Wang, Y.; Xie, Y.; Liu, J. Hepatitis B virus X protein is capable of down-regulating protein level of host antiviral protein APOBEC3G. Sci. Rep. 2017, 7, 40783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Li, H.; Sun, H.; Fan, H.; Hu, Y.; Liu, M.; Li, X.; Tang, H. Hepatitis B Virus-Encoded MicroRNA Controls Viral Replication. J. Virol. 2017, 91, e01919-16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Sun, L.; Mu, T.; Yi, J.; Ma, C.; Xie, H.; Liu, M.; Tang, H. An HBV-encoded miRNA activates innate immunity to restrict HBV replication. J. Mol. Cell Biol. 2020, 12, 263–276. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Jacobs, K.F.K.; Shein, M.; Schütz, A.K.; Mohr, F.; Stadler, H.; Stadler, D.; Lucko, A.M.; Altstetter, S.M.; Wilsch, F.; et al. Efficient and reproducible depletion of hepatitis B virus from plasma derived extracellular vesicles. J. Extracell Vesicles 2020, 10, e12040. [Google Scholar] [CrossRef]
- Dansako, H.; Ueda, Y.; Satoh, S.; Kato, N. Extracellular vesicles activate ATM-Chk2 signaling pathway through the intercellular transfer of mitochondrial DNA in HBV-infected human hepatocytes. FASEB J. 2021, 35, e21680. [Google Scholar] [CrossRef]
- Jung, S.; Altstetter, S.M.; Wilsch, F.; Shein, M.; Schütz, A.K.; Protzer, U. Extracellular vesicles derived from Hepatitis-D Virus infected cells induce a proinflammatory cytokine response in human peripheral blood mononuclear cells and macrophages. Matters 2020, 1–10. [Google Scholar]
- Yao, T.; Lv, M.; Ma, S.; Chen, J.; Zhang, Y.; Yu, Y.; Zang, G.; Chen, X. Ubiquitinated Hepatitis D Antigen-Loaded Microvesicles Induce a Potent Specific Cellular Immune Response to Inhibit HDV Replication in Vivo. Microbiol. Spectr. 2021, 9, e0102421. [Google Scholar] [CrossRef]
- Yang, Y.; Han, Q.; Hou, Z.; Zhang, C.; Tian, Z.; Zhang, J. Exosomes mediate hepatitis B virus (HBV) transmission and NK-cell dysfunction. Cell Mol. Immunol. 2017, 14, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, M.; Inoue, J.; Krueger, E.W.; Chen, J.; Cao, H.; Masamune, A.; McNiven, M.A. The Exosome-Associated Tetraspanin CD63 Contributes to the Efficient Assembly and Infectivity of the Hepatitis B Virus. Hepatol. Commun. 2021, 5, 1238–1251. [Google Scholar] [CrossRef] [PubMed]
- Onódi, Z.; Pelyhe, C.; Terézia Nagy, C.; Brenner, G.B.; Almási, L.; Kittel, Á.; Manček-Keber, M.; Ferdinandy, P.; Buzás, E.I.; Giricz, Z. Isolation of High-Purity Extracellular Vesicles by the Combination of Iodixanol Density Gradient Ultracentrifugation and Bind-Elute Chromatography From Blood Plasma. Front. Physiol. 2018, 9, 1479. [Google Scholar] [CrossRef] [PubMed]
- Willms, E.; Johansson, H.J.; Mäger, I.; Lee, Y.; Blomberg, K.E.; Sadik, M.; Alaarg, A.; Smith, C.I.; Lehtiö, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Glitscher, M.; Tonnemacher, S.; Schollmeier, A.; Raupach, J.; Zahn, T.; Eberle, R.; Krijnse-Locker, J.; Basic, M.; Hildt, E. Presence of Intact Hepatitis B Virions in Exosomes. Cell Mol. Gastroenterol. Hepatol. 2023, 15, 237–259. [Google Scholar] [CrossRef]
- Deng, L.; Jiang, W.; Wang, X.; Merz, A.; Hiet, M.S.; Chen, Y.; Pan, X.; Jiu, Y.; Yang, Y.; Yu, B.; et al. Syntenin regulates hepatitis C virus sensitivity to neutralizing antibody by promoting E2 secretion through exosomes. J. Hepatol. 2019, 71, 52–61. [Google Scholar] [CrossRef]
- Sanada, T.; Hirata, Y.; Naito, Y.; Yamamoto, N.; Kikkawa, Y.; Ishida, Y.; Yamasaki, C.; Tateno, C.; Ochiya, T.; Kohara, M. Transmission of HBV DNA Mediated by Ceramide-Triggered Extracellular Vesicles. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Sukriti, S.; Choudhary, M.C.; Maras, J.S.; Sharma, S.; Thangariyal, S.; Singh, A.; Das, S.; Islam, M.; Sharma, S.; Trehanpati, N.; et al. Extracellular vesicles from hepatitis B patients serve as reservoir of hepatitis B virus DNA. J. Viral Hepat. 2019, 26, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Arzberger, S.; Hösel, M.; Protzer, U. Apoptosis of hepatitis B virus-infected hepatocytes prevents release of infectious virus. J. Virol. 2010, 84, 11994–12001. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walter, A.J.; van de Klundert, M.A.; Jung, S. Many Ways to Communicate—Crosstalk between the HBV-Infected Cell and Its Environment. Pathogens 2023, 12, 29. https://doi.org/10.3390/pathogens12010029
Walter AJ, van de Klundert MA, Jung S. Many Ways to Communicate—Crosstalk between the HBV-Infected Cell and Its Environment. Pathogens. 2023; 12(1):29. https://doi.org/10.3390/pathogens12010029
Chicago/Turabian StyleWalter, Annika Jasmin, Maarten A. van de Klundert, and Stephanie Jung. 2023. "Many Ways to Communicate—Crosstalk between the HBV-Infected Cell and Its Environment" Pathogens 12, no. 1: 29. https://doi.org/10.3390/pathogens12010029