
Group B Streptococcus-Induced Macropinocytosis Contributes to Bacterial Invasion of Brain Endothelial Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
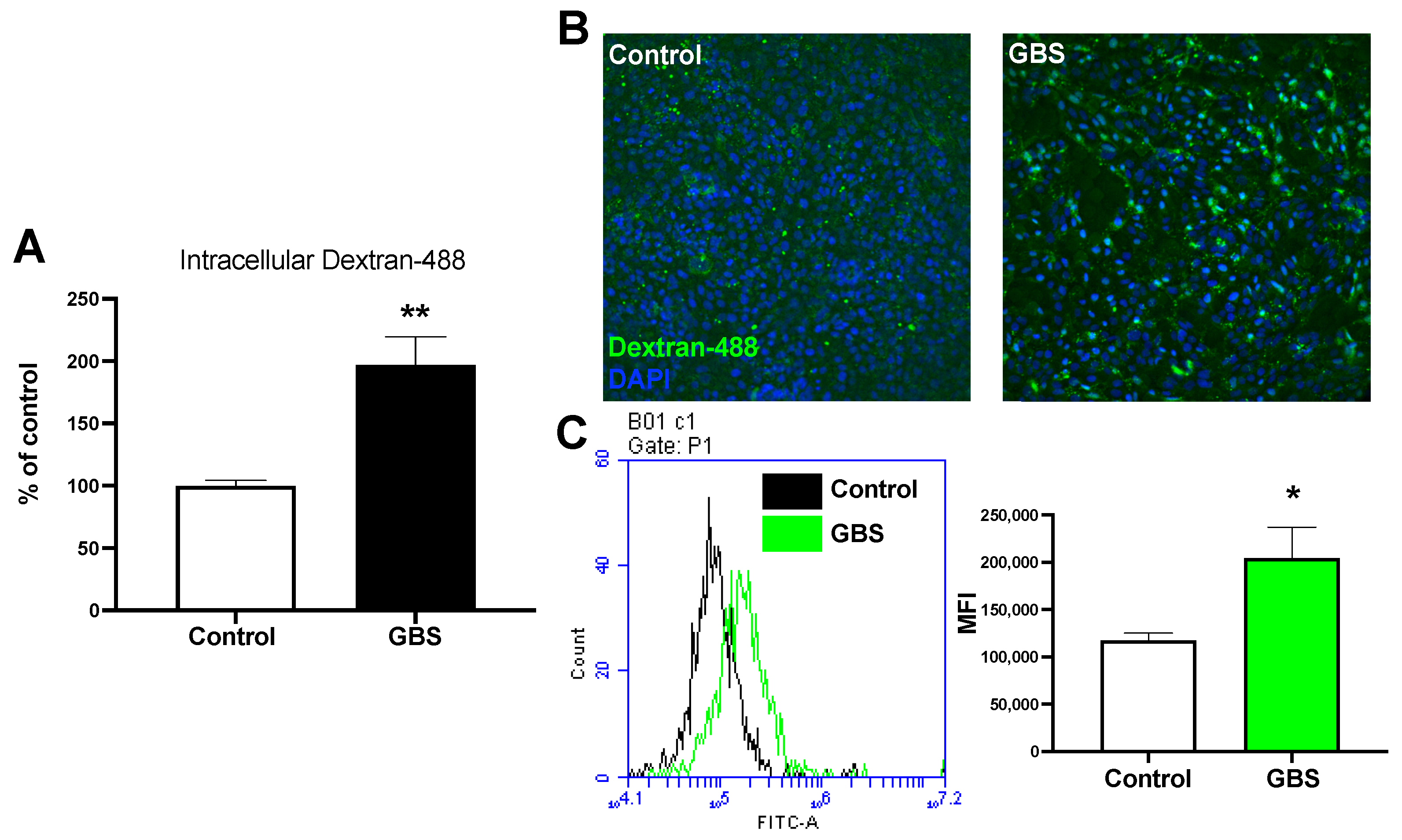
2.1. GBS Infection Increases Rates of Endocytosis
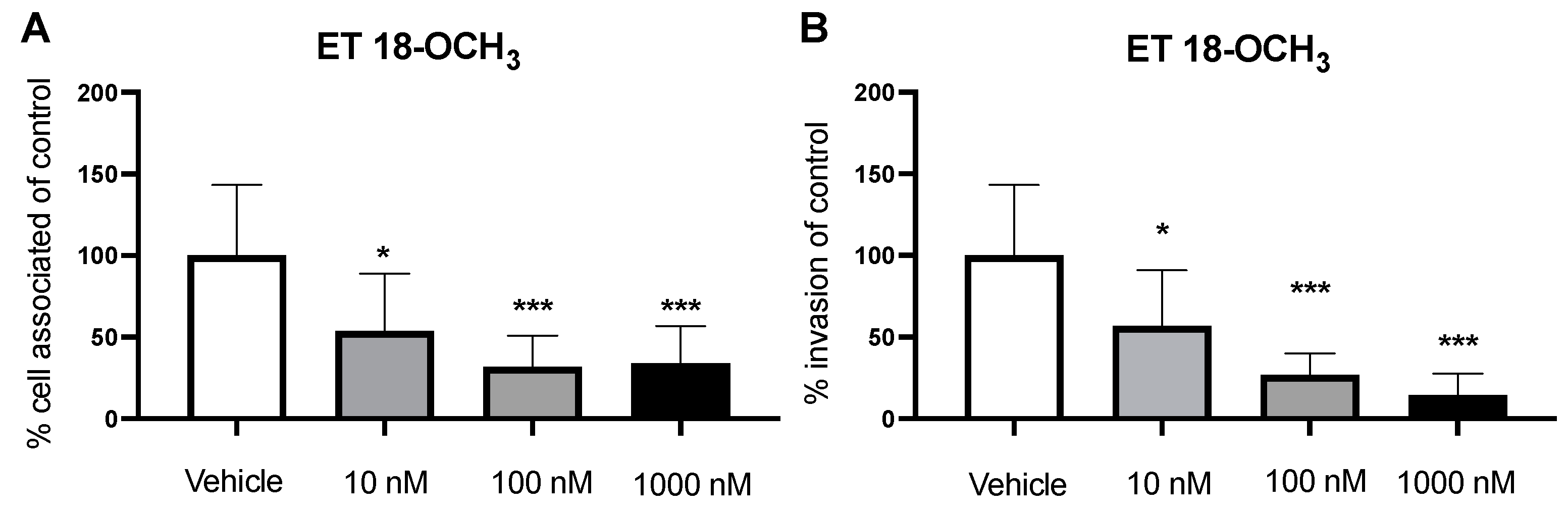
2.2. Inhibition of PLC Decreases GBS Invasion
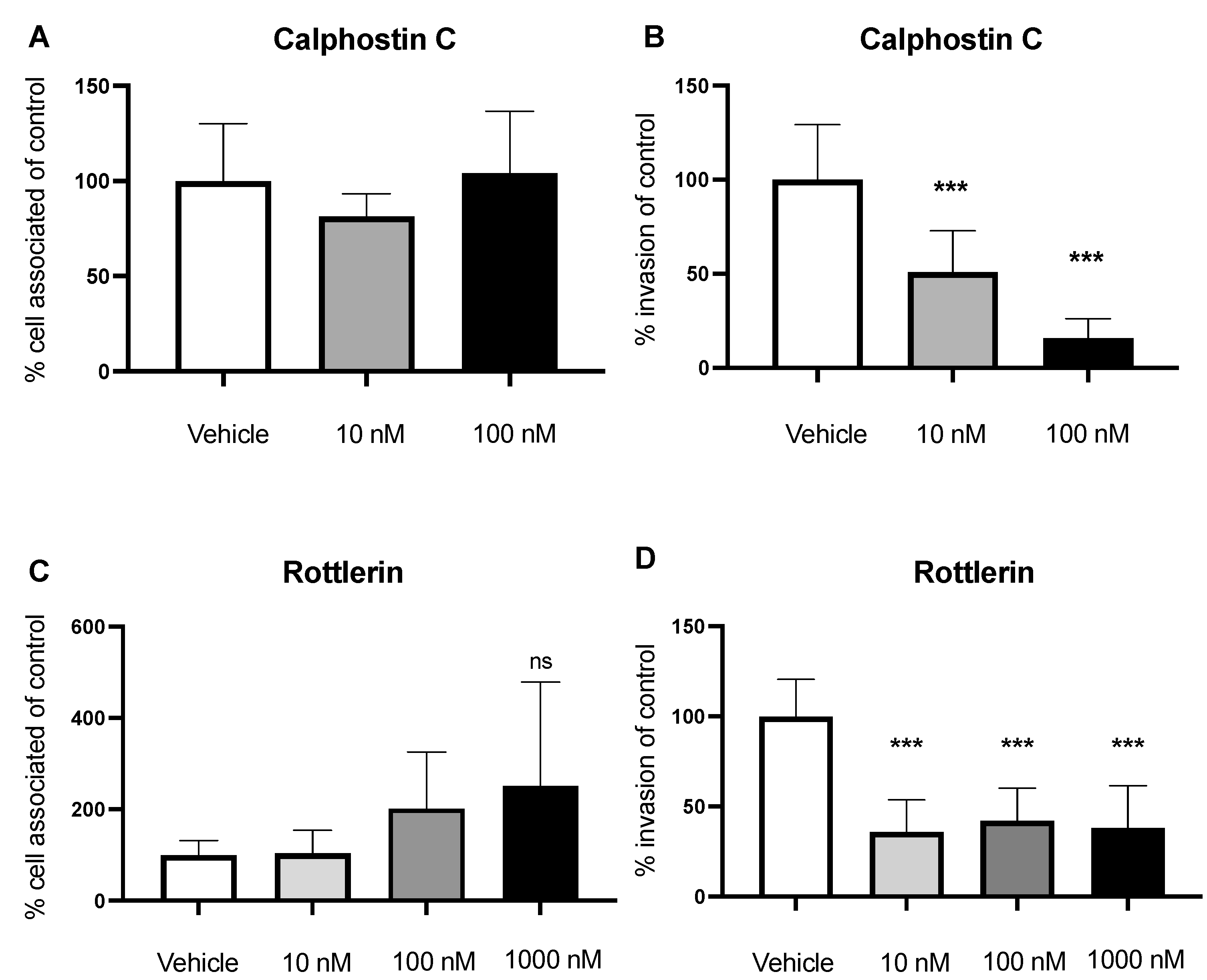
2.3. Inhibition of PKC Decreases Invasion of GBS
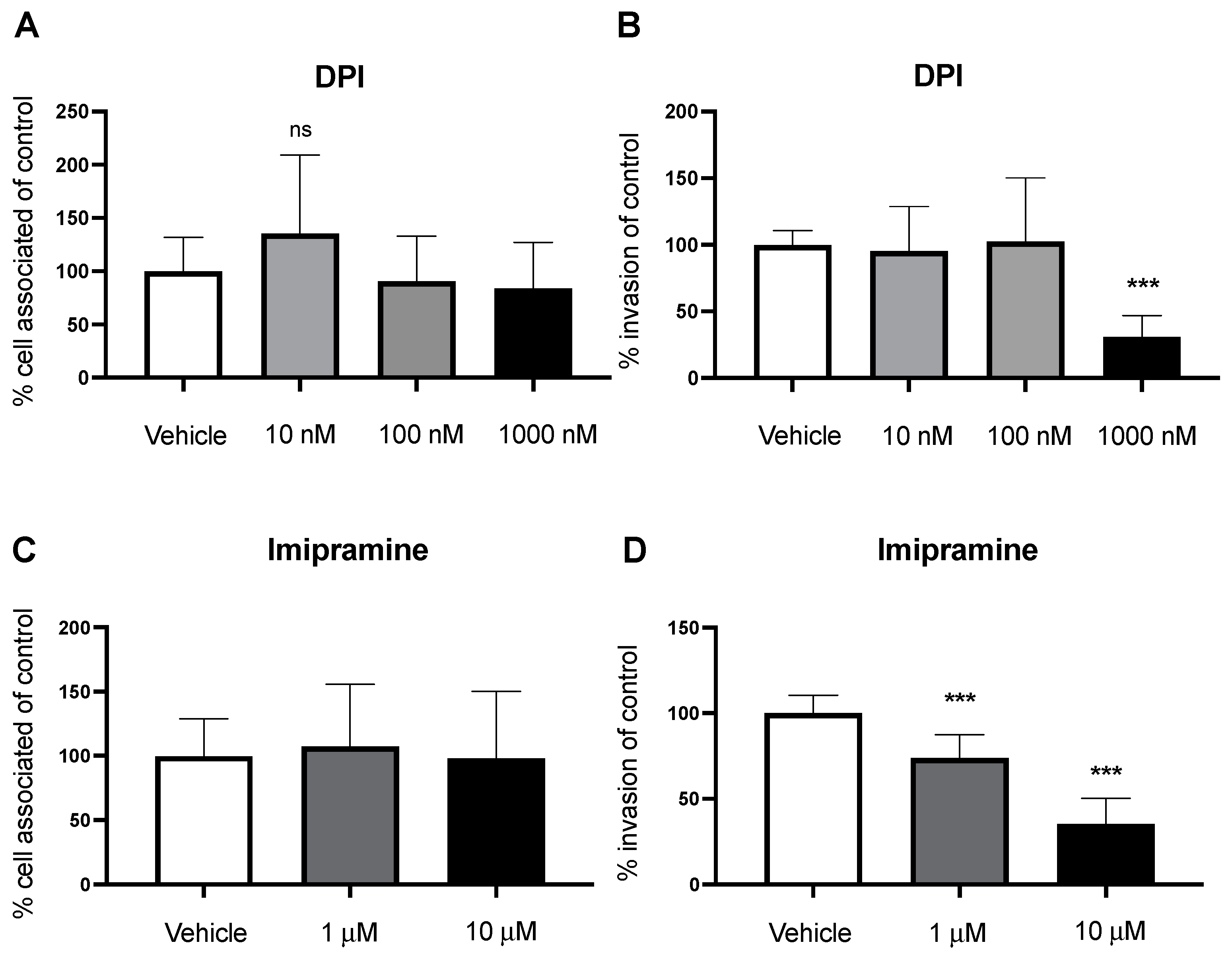
2.4. Nox2 Contributes to GBS Invasion of BECs
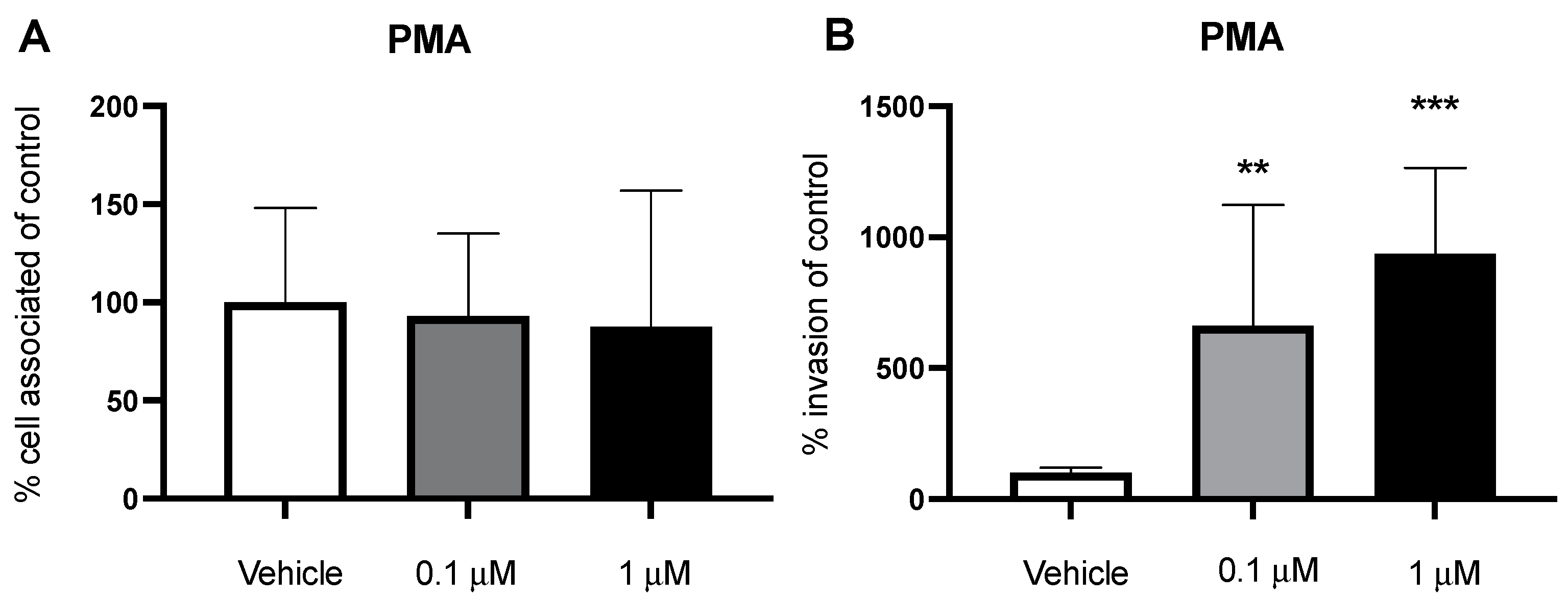
2.5. PMA Stimulates Macropinocytosis and Increases Bacterial Invasion
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Cell Lines
4.2. Brain Endothelial-Cell Differentiation
4.3. Bacterial Preparation and Infection Assays
4.4. Flow Cytometry
4.5. Inhibitors and Antibodies
4.6. Live/Dead Assays
4.7. Growth Curves
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zunt, J.R.; Kassebaum, N.J.; Blake, N.; Glennie, L.; Wright, C.; Nichols, E.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Adamu, A.A.; et al. Global, regional, and national burden of meningitis, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 1061–1082. [Google Scholar] [CrossRef] [Green Version]
- Lucas, L.J.; Brouwer, M.C.; van de Beek, D. Neurological sequelae of bacterial meningitis. J. Infect. 2016, 73, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Doran, K.S.; Fulde, M.; Gratz, N.; Kim, B.J.; Nau, R.; Prasadarao, N.; Schubert-Unkmeir, A.; Tuomanen, E.I.; Valentin-Weigand, P. Host–pathogen interactions in bacterial meningitis. Acta Neuropathol. 2016, 131, 185–209. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J.; McDonagh, M.A.; Deng, L.; Gastfriend, B.D.; Schubert-Unkmeir, A.; Doran, K.S.; Shusta, E.V. Streptococcus agalactiae disrupts P-glycoprotein function in brain endothelial cells. Fluids Barriers CNS 2019, 16, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doran, K.S.; Nizet, V. Molecular pathogenesis of neonatal group B streptococcal infection: No longer in its infancy. Mol. Microbiol. 2004, 54, 23–31. [Google Scholar] [CrossRef]
- Maisey, H.C.; Hensler, M.; Nizet, V.; Doran, K.S. Group B streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. J. Bacteriol. 2007, 189, 1464–1467. [Google Scholar] [CrossRef] [Green Version]
- Doran, K.S.; Engelson, E.J.; Khosravi, A.; Maisey, H.C.; Fedtke, I.; Equils, O.; Michelsen, K.S.; Arditi, M.; Peschel, A.; Nizet, V. Blood-brain barrier invasion by group B Streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. J. Clin. Investig. 2005, 115, 2499–2507. [Google Scholar] [CrossRef] [Green Version]
- Rubin, L.L.; Staddon, J.M. The cell biology of the blood-brain barrier. Annu. Rev. Neurosci. 1999, 22, 11–28. [Google Scholar] [CrossRef]
- Kim, B.J.; Bee, O.B.; Mcdonagh, M.A.; Stebbins, M.J.; Palecek, S.P.; Doran, K.S.; Shusta, E.V. Modeling group B streptococcus and blood-brain barrier interaction by using induced pluripotent stem cell-derived brain endothelial cells. mSphere 2017, 2, e00398-17. [Google Scholar] [CrossRef] [Green Version]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood–brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef]
- Nizet, V.; Kim, K.S.; Stins, M.; Jonas, M.; Chi, E.Y.; Nguyen, D.; Rubens, C.E. Invasion of brain microvascular endothelial cells by group B streptococci. Infect. Immun. 1997, 65, 5074–5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoner, T.D.; Weston, T.A.; Trejo, J.; Doran, K.S. Group B streptococcal infection and activation of human astrocytes. PLoS ONE 2015, 10, e0128431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.P.; Gleeson, P.A. Macropinocytosis: An endocytic pathway for internalising large gulps. Immunol. Cell Biol. 2011, 89, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Loh, L.N.; Mccarthy, E.M.C.; Narang, P.; Khan, N.A.; Ward, T.H. Escherichia coliK1 utilizes host macropinocytic pathways for invasion of brain microvascular endothelial cells. Traffic 2017, 18, 733–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrias, E.S.; Reignault, L.C.; De Souza, W.; Carvalho, T.M. Trypanosoma cruzi uses macropinocytosis as an additional entry pathway into mammalian host cell. Microbes Infect. 2012, 14, 1340–1351. [Google Scholar] [CrossRef]
- Yoshida, S.; Gaeta, I.; Pacitto, R.; Krienke, L.; Alge, O.; Gregorka, B.; Swanson, J.A. Differential signaling during macropinocytosis in response to M-CSF and PMA in macrophages. Front. Physiol. 2015, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Singla, B.; Ghoshal, P.; Lin, H.; Wei, Q.; Dong, Z.; Csányi, G. PKCδ-mediated Nox2 activation promotes fluid-phase pinocytosis of antigens by immature dendritic cells. Front. Immunol. 2018, 9, 537. [Google Scholar] [CrossRef] [Green Version]
- Ghoshal, P.; Singla, B.; Lin, H.; Feck, D.M.; Cantu-Medellin, N.; Kelley, E.E.; Haigh, S.; Fulton, D.; Csányi, G. Nox2-mediated PI3K and cofilin activation confers alternate redox control of macrophage pinocytosis. Antioxid. Redox Signal. 2017, 26, 902–916. [Google Scholar] [CrossRef]
- Lippmann, E.S.; Azarin, S.M.; Kay, J.E.; Nessler, R.A.; Wilson, H.K.; Al-Ahmad, A.; Palecek, S.P.; Shusta, E.V. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 2012, 30, 783–791. [Google Scholar] [CrossRef]
- Stebbins, M.J.; Wilson, H.K.; Canfield, S.G.; Qian, T.; Palecek, S.P.; Shusta, E.V. Differentiation and characterization of human pluripotent stem cell-derived brain microvascular endothelial cells. Methods 2016, 101, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Alimonti, J.B.; Ribecco-Lutkiewicz, M.; Sodja, C.; Jezierski, A.; Stanimirovic, D.B.; Liu, Q.; Haqqani, A.S.; Conlan, W.; Bani-Yaghoub, M. Zika virus crosses an in vitro human blood brain barrier model. Fluids Barriers CNS 2018, 15, 15. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Haferkamp, U.; Pfefferle, S.; Woo, M.S.; Heinrich, F.; Schweizer, M.; Appelt-Menzel, A.; Cubukova, A.; Barenberg, J.; Leu, J.; et al. The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem Cell Rep. 2022, 17, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Schneider, V.A.F.; Coorens, M.; Tjeerdsma-van Bokhoven, J.L.M.; Posthuma, G.; van Dijk, A.; Veldhuizen, E.J.A.; Haagsman, H.P. Imaging the antistaphylococcal activity of CATH-2: Mechanism of attack and regulation of inflammatory response. mSphere 2017, 2, e00370-17. [Google Scholar] [CrossRef] [Green Version]
- Martins Gomes, S.F.; Westermann, A.J.; Sauerwein, T.; Hertlein, T.; Förstner, K.U.; Ohlsen, K.; Metzger, M.; Shusta, E.V.; Kim, B.J.; Appelt-Menzel, A.; et al. Induced pluripotent stem cell-derived brain endothelial cells as a cellular model to study neisseria meningitidis infection. Front. Microbiol. 2019, 10, 1181. [Google Scholar] [CrossRef] [PubMed]
- Endres, L.M.; Schubert-Unkmeir, A.; Kim, B.J. Neisseria meningitidis infection of induced pluripotent stem-cell derived brain endothelial cells. J. Vis. Exp. 2020, 161, e61400. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Hancock, B.M.; Bermudez, A.; Cid, N.D.; Reyes, E.; Van Sorge, N.M.; Lauth, X.; Smurthwaite, C.A.; Hilton, B.J.; Stotland, A.; et al. Bacterial induction of Snail1 contributes to blood-brain barrier disruption. J. Clin. Investig. 2015, 125, 2473–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, M.P. PIP2 and PIP3. Cell 2000, 100, 603–606. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.; Fabian, L.; Forer, A.; Brill, J.A. Phospholipase C and myosin light chain kinase inhibition define a common step in actin regulation during cytokinesis. BMC Cell Biol. 2007, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- El-Benna, J.; Dang, P.M.C.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef]
- Lin, H.P.; Singla, B.; Ghoshal, P.; Faulkner, J.L.; Cherian-Shaw, M.; O’Connor, P.M.; She, J.X.; Belin De Chantemele, E.J.; Csányi, G. Identification of novel macropinocytosis inhibitors using a rational screen of Food and Drug Administration-approved drugs. Br. J. Pharmacol. 2018, 175, 3640–3655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutting, A.S.; Del Rosario, Y.; Mu, R.; Rodriguez, A.; Till, A.; Subramani, S.; Gottlieb, R.A.; Doran, K.S. The role of autophagy during group B Streptococcus infection of blood-brain barrier endothelium. J. Biol. Chem. 2014, 289, 35711–35723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruvada, R.; Zhu, L.; Pearce, D.; Sapirstein, A.; Kim, K.S. Host cytosolic phospholipase A2α contributes to group B Streptococcus penetration of the blood-brain barrier. Infect Immun. 2011, 79, 4088–4093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, C.; Prakash, A.; Hou, Z.; Liu, W.; She, W.; Morris, A.; Sik Kim, K. Therapeutic development of group B Streptococcus meningitis by targeting a host cell signaling network involving EGFR. EMBO Mol. Med. 2021, 13, e12651. [Google Scholar] [CrossRef] [PubMed]
- de Cambronne, R.D.; Fouet, A.; Picart, A.; Bourrel, A.S.; Anjou, C.; Bouvier, G.; Candeias, C.; Bouaboud, A.; Costa, L.; Boulay, A.C.; et al. CC17 group B Streptococcus exploits integrins for neonatal meningitis development. J. Clin. Investig. 2021, 131, e136737. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.S.; Minasov, G.; Seepersaud, R.; Doran, K.S.; Dubrovska, I.; Shuvalova, L.; Anderson, W.F.; Iverson, T.M.; Sullam, P.M. Characterization of fibrinogen binding by glycoproteins Srr1 and Srr2 of Streptococcus agalactiae. J. Biol. Chem. 2013, 288, 35982–35996. [Google Scholar] [CrossRef] [Green Version]
- Shabayek, S.; Spellerberg, B. Group B Streptococcal colonization, molecular characteristics, and epidemiology. Front. Microbiol. 2018, 9, 437. [Google Scholar] [CrossRef]
- Kumari, S.; Mg, S.; Mayor, S. Endocytosis unplugged: Multiple ways to enter the cell. Cell Res. 2010, 20, 256–275. [Google Scholar] [CrossRef] [Green Version]
- Lemire, P.; Houde, M.; Segura, M. Encapsulated group B Streptococcus modulates dendritic cell functions via lipid rafts and clathrin-mediated endocytosis. Cell Microbiol. 2012, 14, 1707–1719. [Google Scholar] [CrossRef]
- Goluszko, P.; Popov, V.; Wen, J.; Jones, A.; Yallampalli, C. Group B streptococcus exploits lipid rafts and phosphoinositide 3-kinase/Akt signaling pathway to invade human endometrial cells. Am. J. Obstet. Gynecol. 2008, 199, 548. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood—brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Rubens, C.E.; Wessels, M.R.; Heggen, L.M.; Kasper, D.L. Transposon mutagenesis of type III group B Streptococcus: Correlation of capsule expression with virulence. Proc. Natl. Acad. Sci. USA 1987, 84, 7208–7212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Kim, B.J.; Carmona, E.M.; Cutting, A.S.; Gurney, M.A.; Carlos, C.; Feuer, R.; Prasadarao, N.V.; Doran, K.S. Bacterial Pili exploit integrin machinery to promote immune activation and efficient blood-brain barrier penetration. Nat. Commun. 2011, 2, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espinal, E.R.; Matthews, T.; Holder, B.M.; Bee, O.B.; Humber, G.M.; Brook, C.E.; Divyapicigil, M.; Sharp, J.; Kim, B.J. Group B Streptococcus-Induced Macropinocytosis Contributes to Bacterial Invasion of Brain Endothelial Cells. Pathogens 2022, 11, 474. https://doi.org/10.3390/pathogens11040474
Espinal ER, Matthews T, Holder BM, Bee OB, Humber GM, Brook CE, Divyapicigil M, Sharp J, Kim BJ. Group B Streptococcus-Induced Macropinocytosis Contributes to Bacterial Invasion of Brain Endothelial Cells. Pathogens. 2022; 11(4):474. https://doi.org/10.3390/pathogens11040474
Chicago/Turabian StyleEspinal, Eric R., Teralan Matthews, Brianna M. Holder, Olivia B. Bee, Gabrielle M. Humber, Caroline E. Brook, Mustafa Divyapicigil, Jerod Sharp, and Brandon J. Kim. 2022. "Group B Streptococcus-Induced Macropinocytosis Contributes to Bacterial Invasion of Brain Endothelial Cells" Pathogens 11, no. 4: 474. https://doi.org/10.3390/pathogens11040474