
Clinical and Molecular Basis of Hepatocellular Carcinoma after Hepatitis C Virus Eradication


{kind=link}
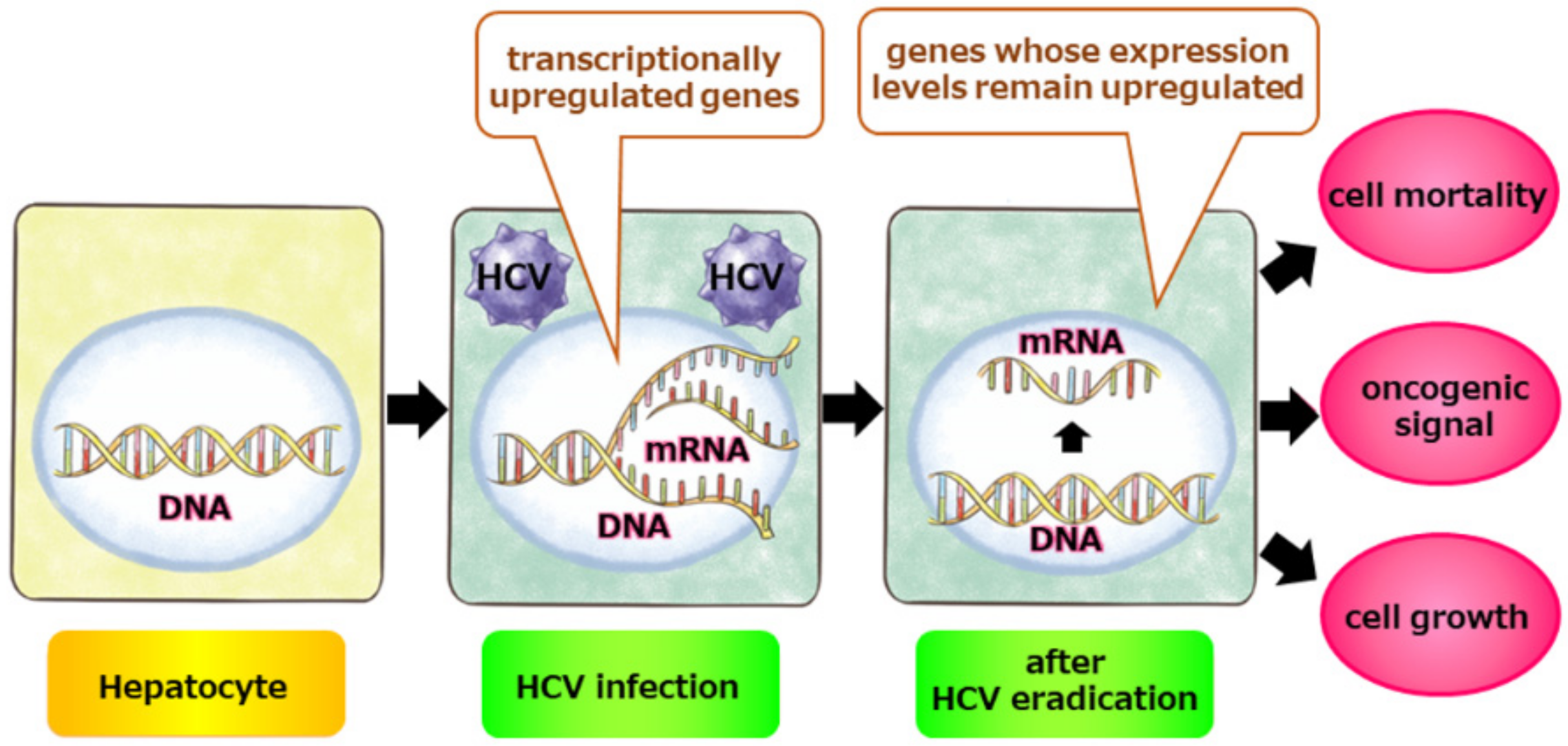{kind=link}
Abstract
:1. Introduction
2. Progress of Anti-HCV Therapy and Its Protective Effect against Hepatocarcinogenesis
3. Suppression of HCC Development by DAA Therapy
4. Risk Factors for HCC Development after HCV Eradication Using DAA Therapy
5. Molecular Basis of Post-SVR Hepatocarcinogenesis: Genetic Alterations
6. Molecular Basis for Post-SVR Hepatocarcinogenesis: Epigenetic Alterations
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, C.J. Liver cancer. In World Cancer Report 2020; Stewart, B.W., Weiderpass, E., Wild, C.P., Eds.; International Agency for Research on Cancer: Lyon, France, 2020; p. 355. [Google Scholar]
- European Association for The Study of The Liver. EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiha, G.; Mousa, N.; Soliman, R.; Nnh Mikhail, N.; Adel Elbasiony, M.; Khattab, M. Incidence of HCC in chronic hepatitis C patients with advanced hepatic fibrosis who achieved SVR following DAAs: A prospective study. J. Viral. Hepat. 2020, 27, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients with Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, G.N.; Green, P.K.; Berry, K. HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. J. Hepatol. 2017, 17, S0168–S8278. [Google Scholar] [CrossRef]
- Calvaruso, V.; Cabibbo, G.; Cacciola, I.; Petta, S.; Madonia, S.; Bellia, A.; Tinè, F.; Distefano, M.; Licata, A.; Giannitrapani, L.; et al. Incidence of Hepatocellular Carcinoma in Patients with HCV-Associated Cirrhosis Treated with Direct-Acting Antiviral Agents. Gastroenterology 2018, 155, 411–421.e414. [Google Scholar] [CrossRef] [Green Version]
- Pawlotsky, J.M.; Feld, J.J.; Zeuzem, S.; Hoofnagle, J.H. From non-A, non-B hepatitis to hepatitis C virus cure. J. Hepatol. 2015, 62, S87–S99. [Google Scholar] [CrossRef] [Green Version]
- McHutchison, J.G.; Gordon, S.C.; Schiff, E.R.; Shiffman, M.L.; Lee, W.M.; Rustgi, V.K.; Goodman, Z.D.; Ling, M.H.; Cort, S.; Albrecht, J.K.; et al. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N. Engl. J. Med. 1998, 339, 1485–1492. [Google Scholar] [CrossRef] [Green Version]
- Poynard, T.; Marcellin, P.; Lee, S.S.; Niederau, C.; Minuk, G.S.; Ideo, G.; Bain, V.; Heathcote, J.; Zeuzem, S.; Trepo, C.; et al. Randomised trial of interferon alpha2b plus ribavirin for 48 weeks or for 24 weeks versus interferon alpha2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. International Hepatitis Interventional Therapy Group (IHIT). Lancet 1998, 352, 1426–1432. [Google Scholar] [CrossRef]
- Di Bisceglie, A.M.; Shindo, M.; Fong, T.L.; Fried, M.W.; Swain, M.G.; Bergasa, N.V.; Axiotis, C.A.; Waggoner, J.G.; Park, Y.; Hoofnagle, J.H. A pilot study of ribavirin therapy for chronic hepatitis C. Hepatology 1992, 16, 649–654. [Google Scholar] [CrossRef]
- Manns, M.P.; McHutchison, J.G.; Gordon, S.C.; Rustgi, V.K.; Shiffman, M.; Reindollar, R.; Goodman, Z.D.; Koury, K.; Ling, M.; Albrecht, J.K.; et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet 2001, 358, 958–965. [Google Scholar] [CrossRef]
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Gonçales, F.L., Jr.; Häussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 2002, 347, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Ghany, M.G.; Nelson, D.R.; Strader, D.B.; Thomas, D.L.; Seeff, L.B. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011, 54, 1433–1444. [Google Scholar] [CrossRef] [Green Version]
- Sarrazin, C. Treatment failure with DAA therapy: Importance of resistance. J. Hepatol. 2021, 74, 1472–1482. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Recommendations on Treatment of Hepatitis C 2018. J. Hepatol. 2018, 69, 461–511. [Google Scholar] [CrossRef] [Green Version]
- Asahina, Y. JSH Guidelines for the Management of Hepatitis C Virus Infection, 2019 Update; Protective Effect of Antiviral Therapy against Hepatocarcinogenesis. Hepatol. Res. 2020, 50, 775–790. [Google Scholar] [CrossRef]
- Ikeda, K.; Saitoh, S.; Arase, Y.; Chayama, K.; Suzuki, Y.; Kobayashi, M.; Tsubota, A.; Nakamura, I.; Murashima, N.; Kumada, H.; et al. Effect of interferon therapy on hepatocellular carcinogenesis in patients with chronic hepatitis type C: A long-term observation study of 1643 patients using statistical bias correction with proportional hazard analysis. Hepatology 1999, 29, 1124–1130. [Google Scholar] [CrossRef]
- Nishiguchi, S.; Kuroki, T.; Nakatani, S.; Morimoto, H.; Takeda, T.; Nakajima, S.; Shiomi, S.; Seki, S.; Kobayashi, K.; Otani, S. Randomised trial of effects of interferon-alpha on incidence of hepatocellular carcinoma in chronic active hepatitis C with cirrhosis. Lancet 1995, 346, 1051–1055. [Google Scholar] [CrossRef]
- Conti, F.; Buonfiglioli, F.; Scuteri, A.; Crespi, C.; Bolondi, L.; Caraceni, P.; Foschi, F.G.; Lenzi, M.; Mazzella, G.; Verucchi, G.; et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016, 65, 727–733. [Google Scholar] [CrossRef]
- Ravi, S.; Axley, P.; Jones, D.; Kodali, S.; Simpson, H.; McGuire, B.M.; Singal, A.K. Unusually High Rates of Hepatocellular Carcinoma after Treatment with Direct-Acting Antiviral Therapy for Hepatitis C Related Cirrhosis. Gastroenterology 2017, 152, 911–912. [Google Scholar] [CrossRef] [Green Version]
- Waziry, R.; Hajarizadeh, B.; Grebely, J.; Amin, J.; Law, M.; Danta, M.; George, J.; Dore, G.J. Hepatocellular carcinoma risk following direct-acting antiviral HCV therapy: A systematic review, meta-analyses, and meta-regression. J. Hepatol. 2017, 67, 1204–1212. [Google Scholar] [CrossRef]
- Huang, P.; Liu, M.; Zang, F.; Yao, Y.; Yue, M.; Wang, J.; Fan, H.; Zhuo, L.; Wu, J.; Xia, X.; et al. The development of hepatocellular carcinoma in HCV-infected patients treated with DAA: A comprehensive analysis. Carcinogenesis 2018, 39, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Han, D.H.; Shin, H.J.; Lee, J.S.; Kim, S.U.; Park, J.Y.; Kim, D.Y.; Ahn, S.H.; Kim, B.K. Hepatocellular Carcinoma Risk According to Regimens for Eradication of Hepatitis C Virus; Interferon or Direct Acting Antivirals. Cancers 2020, 12, 3414. [Google Scholar] [CrossRef] [PubMed]
- Li, D.K.; Ren, Y.; Fierer, D.S.; Rutledge, S.; Shaikh, O.S.; Lo Re, V., 3rd; Simon, T.; Abou-Samra, A.; Chung, R.T.; Butt, A.A. The short-term incidence of hepatocellular carcinoma is not increased after hepatitis C treatment with direct-acting antivirals: An ERCHIVES study. Hepatology 2018, 67, 2244–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated with Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005.e1001. [Google Scholar] [CrossRef] [Green Version]
- Jilkova, Z.M.; Saleem, K.; Afzal, S.; Decaens, T. Predictive Factors for Hepatocellular Carcinoma Development after Direct-Acting Antiviral Treatment of HCV. Livers 2021, 1, 313–321. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Beste, L.A.; Green, P.K.; Singal, A.G.; Tapper, E.B.; Waljee, A.K.; Sterling, R.K.; Feld, J.J.; Kaplan, D.E.; Taddei, T.H.; et al. Increased Risk for Hepatocellular Carcinoma Persists up to 10 Years after HCV Eradication in Patients with Baseline Cirrhosis or High FIB-4 Scores. Gastroenterology 2019, 157, 1264–1278.e1264. [Google Scholar] [CrossRef] [Green Version]
- Pons, M.; Rodríguez-Tajes, S.; Esteban, J.I.; Mariño, Z.; Vargas, V.; Lens, S.; Buti, M.; Augustin, S.; Forns, X.; Mínguez, B.; et al. Non-invasive prediction of liver-related events in patients with HCV-associated compensated advanced chronic liver disease after oral antivirals. J. Hepatol. 2020, 72, 472–480. [Google Scholar] [CrossRef]
- You, M.W.; Kim, K.W.; Shim, J.J.; Pyo, J. Impact of liver-stiffness measurement on hepatocellular carcinoma development in chronic hepatitis C patients treated with direct-acting antivirals: A systematic review and time-to-event meta-analysis. J. Gastroenterol. Hepatol. 2021, 36, 601–608. [Google Scholar] [CrossRef]
- Krassenburg, L.A.P.; Maan, R.; Ramji, A.; Manns, M.P.; Cornberg, M.; Wedemeyer, H.; de Knegt, R.J.; Hansen, B.E.; Janssen, H.L.A.; Man, R.A.; et al. Clinical outcomes following DAA therapy in patients with HCV-related cirrhosis depend on disease severity. J. Hepatol. 2021, 74, 1053–1063. [Google Scholar] [CrossRef]
- Sangiovanni, A.; Alimenti, E.; Gattai, R.; Filomia, R.; Parente, E.; Valenti, L.; Marzi, L.; Pellegatta, G.; Borgia, G.; Gambato, M.; et al. Undefined/non-malignant hepatic nodules are associated with early occurrence of HCC in DAA-treated patients with HCV-related cirrhosis. J. Hepatol. 2020, 73, 593–602. [Google Scholar] [CrossRef]
- Mashiba, T.; Joko, K.; Kurosaki, M.; Ochi, H.; Osaki, Y.; Kojima, Y.; Nakata, R.; Goto, T.; Takehiro, A.; Kimura, H.; et al. Does interferon-free direct-acting antiviral therapy for hepatitis C after curative treatment for hepatocellular carcinoma lead to unexpected recurrences of HCC? A multicenter study by the Japanese Red Cross Hospital Liver Study Group. PLoS ONE 2018, 13, e0194704. [Google Scholar] [CrossRef]
- Cabibbo, G.; Celsa, C.; Calvaruso, V.; Petta, S.; Cacciola, I.; Cannavò, M.R.; Madonia, S.; Rossi, M.; Magro, B.; Rini, F.; et al. Direct-acting antivirals after successful treatment of early hepatocellular carcinoma improve survival in HCV-cirrhotic patients. J. Hepatol. 2019, 71, 265–273. [Google Scholar] [CrossRef]
- Singal, A.G.; Rich, N.E.; Mehta, N.; Branch, A.D.; Pillai, A.; Hoteit, M.; Volk, M.; Odewole, M.; Scaglione, S.; Guy, J.; et al. Direct-Acting Antiviral Therapy for Hepatitis C Virus Infection Is Associated with Increased Survival in Patients with a History of Hepatocellular Carcinoma. Gastroenterology 2019, 157, 1253–1263.e1252. [Google Scholar] [CrossRef] [Green Version]
- Dang, H.; Yeo, Y.H.; Yasuda, S.; Huang, C.F.; Iio, E.; Landis, C.; Jun, D.W.; Enomoto, M.; Ogawa, E.; Tsai, P.C.; et al. Cure With Interferon-Free Direct-Acting Antiviral Is Associated with Increased Survival in Patients with Hepatitis C Virus-Related Hepatocellular Carcinoma from Both East and West. Hepatology 2020, 71, 1910–1922. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Cusi, K.; Pettiti, M.; Hardies, J.; Miyazaki, Y.; Berria, R.; Buzzigoli, E.; Sironi, A.M.; Cersosimo, E.; Ferrannini, E.; et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007, 133, 496–506. [Google Scholar] [CrossRef]
- Siphepho, P.Y.; Liu, Y.T.; Shabangu, C.S.; Huang, J.F.; Huang, C.F.; Yeh, M.L.; Yu, M.L.; Wang, S.C. The Impact of Steatosis on Chronic Hepatitis C Progression and Response to Antiviral Treatments. Biomedicines 2021, 9, 1491. [Google Scholar] [CrossRef]
- Ji, D.; Chen, G.F.; Niu, X.X.; Zhang, M.; Wang, C.; Shao, Q.; Wu, V.; Wang, Y.; Cheng, G.; Hurwitz, S.J.; et al. Non-alcoholic fatty liver disease is a risk factor for occurrence of hepatocellular carcinoma after sustained virologic response in chronic hepatitis C patients: A prospective four-years follow-up study. Metabol. Open 2021, 10, 100090. [Google Scholar] [CrossRef]
- Minami, T.; Tateishi, R.; Fujiwara, N.; Nakagomi, R.; Nakatsuka, T.; Sato, M.; Uchino, K.; Enooku, K.; Nakagawa, H.; Fujinaga, H.; et al. Impact of Obesity and Heavy Alcohol Consumption on Hepatocellular Carcinoma Development after HCV Eradication with Antivirals. Liver Cancer 2021, 10, 309–319. [Google Scholar] [CrossRef]
- Ogawa, E.; Nomura, H.; Nakamuta, M.; Furusyo, N.; Kajiwara, E.; Dohmen, K.; Kawano, A.; Ooho, A.; Azuma, K.; Takahashi, K.; et al. Development of Hepatocellular Carcinoma by Patients Aged 75-84 with Chronic Hepatitis C Treated with Direct-Acting Antivirals. J. Infect. Dis. 2020, jiaa359. [Google Scholar] [CrossRef]
- Yoshimasu, Y.; Furuichi, Y.; Kasai, Y.; Takeuchi, H.; Sugimoto, K.; Nakamura, I.; Itoi, T. Predictive factors for hepatocellular carcinoma occurrence or recurrence after direct-acting antiviral agents in patients with chronic hepatitis C. J. Gastrointestin. Liver Dis. 2019, 28, 63–71. [Google Scholar] [CrossRef]
- Debes, J.D.; van Tilborg, M.; Groothuismink, Z.M.A.; Hansen, B.E.; Schulze Zur Wiesch, J.; von Felden, J.; de Knegt, R.J.; Boonstra, A. Levels of Cytokines in Serum Associate with Development of Hepatocellular Carcinoma in Patients with HCV Infection Treated with Direct-Acting Antivirals. Gastroenterology 2018, 154, 515–517.e513. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Asch, S.M.; Cao, Y.; Li, L.; El-Serag, H.B. Long-Term Risk of Hepatocellular Carcinoma in HCV Patients Treated with Direct Acting Antiviral Agents. Hepatology 2020, 71, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Syed, T.; Fazili, J.D.; Ali, I.A.; Zhao, D.; Hughes, D.; Mahmood, S.M. Hepatocellular Carcinoma Occurrence and Recurrence in Hepatitis C-Infected Patients Treated with Direct-Acting Antivirals. Cureus 2018, 10, e2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, G.N.; Green, P.K.; Beste, L.A.; Mun, E.J.; Kerr, K.F.; Berry, K. Development of models estimating the risk of hepatocellular carcinoma after antiviral treatment for hepatitis C. J. Hepatol. 2018, 69, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar]
- Nault, J.C.; Calderaro, J.; Di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Yamamoto, S.; Arai, Y.; Hosoda, F.; Ishikawa, S.; Tsutsumi, S.; Sonoda, K.; Totsuka, H.; Shirakihara, T.; et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat. Genet. 2011, 43, 464–469. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e1224. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Takai, A.; Eso, Y.; Takahashi, K.; Marusawa, H.; Seno, H. Genetic Landscape of Multistep Hepatocarcinogenesis. Cancers 2022, 14, 568. [Google Scholar] [CrossRef]
- Ikeda, A.; Shimizu, T.; Matsumoto, Y.; Fujii, Y.; Eso, Y.; Inuzuka, T.; Mizuguchi, A.; Shimizu, K.; Hatano, E.; Uemoto, S.; et al. Leptin receptor somatic mutations are frequent in HCV-infected cirrhotic liver and associated with hepatocellular carcinoma. Gastroenterology 2014, 146, 222–232.e235. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Takeda, H.; Takai, A.; Matsumoto, T.; Kakiuchi, N.; Yokoyama, A.; Yoshida, K.; Kaido, T.; Uemoto, S.; Minamiguchi, S.; et al. Comprehensive analysis of genetic aberrations linked to tumorigenesis in regenerative nodules of liver cirrhosis. J. Gastroenterol. 2019, 54, 628–640. [Google Scholar] [CrossRef]
- Brunner, S.F.; Roberts, N.D.; Wylie, L.A.; Moore, L.; Aitken, S.J.; Davies, S.E.; Sanders, M.A.; Ellis, P.; Alder, C.; Hooks, Y.; et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019, 574, 538–542. [Google Scholar] [CrossRef]
- Zhu, M.; Lu, T.; Jia, Y.; Luo, X.; Gopal, P.; Li, L.; Odewole, M.; Renteria, V.; Singal, A.G.; Jang, Y.; et al. Somatic Mutations Increase Hepatic Clonal Fitness and Regeneration in Chronic Liver Disease. Cell 2019, 177, 608–621.e612. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Takai, A.; Kumagai, K.; Iguchi, E.; Arasawa, S.; Eso, Y.; Shimizu, T.; Ueda, Y.; Taura, K.; Uemoto, S.; et al. Multiregional whole-genome sequencing of hepatocellular carcinoma with nodule-in-nodule appearance reveals stepwise cancer evolution. J. Pathol. 2020, 252, 398–410. [Google Scholar] [CrossRef]
- Müller, M.; Bird, T.G.; Nault, J.C. The landscape of gene mutations in cirrhosis and hepatocellular carcinoma. J. Hepatol. 2020, 72, 990–1002. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, H.J.; Kim, J.H.; Lee, H.S.; Jang, J.J.; Kang, G.H. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am. J. Pathol. 2003, 163, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Nagasaka, T.; Nishimura, T.; Ikai, I.; Boland, C.R.; Goel, A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology 2008, 47, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y.; Shinjo, K.; Shimizu, Y.; Sano, T.; Yamao, K.; Gao, W.; Fujii, M.; Osada, H.; Sekido, Y.; Murakami, S.; et al. Hepatitis virus infection affects DNA methylation in mice with humanized livers. Gastroenterology 2014, 146, 562–572. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Rusyn, I. Role of epigenetic aberrations in the development and progression of human hepatocellular carcinoma. Cancer Lett. 2014, 342, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Hamdane, N.; Jühling, F.; Crouchet, E.; El Saghire, H.; Thumann, C.; Oudot, M.A.; Bandiera, S.; Saviano, A.; Ponsolles, C.; Suarez, A.A.R.; et al. HCV-Induced Epigenetic Changes Associated with Liver Cancer Risk Persist after Sustained Virologic Response. Gastroenterology 2019, 156, 2313–2329.e2317. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Takai, A.; Iguchi, E.; Mishima, M.; Arasawa, S.; Kumagai, K.; Eso, Y.; Shimizu, T.; Takahashi, K.; Ueda, Y.; et al. Oncogenic transcriptomic profile is sustained in the liver after the eradication of the hepatitis C virus. Carcinogenesis 2021, 42, 672–684. [Google Scholar] [CrossRef]
- Cassim, S.; Raymond, V.A.; Dehbidi-Assadzadeh, L.; Lapierre, P.; Bilodeau, M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle 2018, 17, 903–916. [Google Scholar] [CrossRef]
- Aizarani, N.; Saviano, A.; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef]
- Chen, W.T.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991.e919. [Google Scholar] [CrossRef]
- Take, S.; Mizuno, M.; Ishiki, K.; Kusumoto, C.; Imada, T.; Hamada, F.; Yoshida, T.; Yokota, K.; Mitsuhashi, T.; Okada, H. Risk of gastric cancer in the second decade of follow-up after Helicobacter pylori eradication. J. Gastroenterol. 2020, 55, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Marusawa, H.; Matsumoto, Y.; Inuzuka, T.; Ikeda, A.; Fujii, Y.; Minamiguchi, S.; Miyamoto, S.; Kou, T.; Sakai, Y.; et al. Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology 2014, 147, 407–417.e403. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oe, N.; Takeda, H.; Eso, Y.; Takai, A.; Marusawa, H. Clinical and Molecular Basis of Hepatocellular Carcinoma after Hepatitis C Virus Eradication. Pathogens 2022, 11, 430. https://doi.org/10.3390/pathogens11040430
Oe N, Takeda H, Eso Y, Takai A, Marusawa H. Clinical and Molecular Basis of Hepatocellular Carcinoma after Hepatitis C Virus Eradication. Pathogens. 2022; 11(4):430. https://doi.org/10.3390/pathogens11040430
Chicago/Turabian StyleOe, Natsumi, Haruhiko Takeda, Yuji Eso, Atsushi Takai, and Hiroyuki Marusawa. 2022. "Clinical and Molecular Basis of Hepatocellular Carcinoma after Hepatitis C Virus Eradication" Pathogens 11, no. 4: 430. https://doi.org/10.3390/pathogens11040430