
Rational-Based Discovery of Novel β-Carboline Derivatives as Potential Antimalarials: From In Silico Identification of Novel Targets to Inhibition of Experimental Cerebral Malaria
 , and
, and
Abstract
:1. Introduction
2. Experimental Design
2.1. Virtual Screening
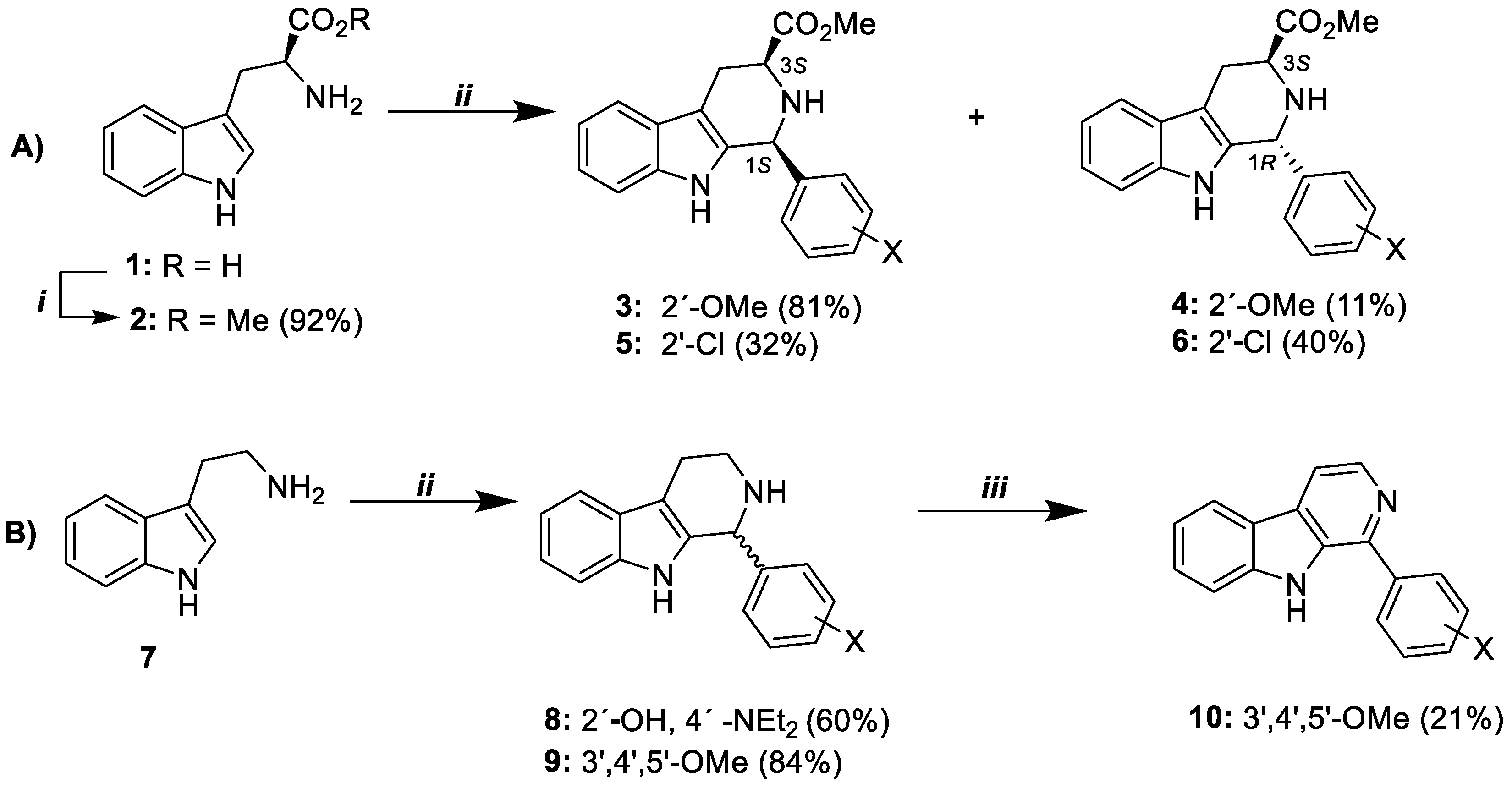
2.2. Chemistry
2.2.1. Synthesis of L-Tryptophan Methyl Ester (2)
2.2.2. General Procedure for the Synthesis of 3, 4, 5, 6, 8, and 9
2.2.3. Methyl (1S,3S)-1-(2-Methoxy)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (3)
2.2.4. Methyl (1R,3S)-1-(2-Methoxy)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (4)
2.2.5. Methyl (1S,3S)-1-(2-Chloro)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (5)
2.2.6. Methyl (1R,3S)-1-(2-Chloro)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (6)
2.2.7. 5-(Diethylamino)-2-(2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)phenol (8)
2.2.8. 1-(3,4,5-Trimethoxyphenyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (9)
2.2.9. 1-(3,4,5-Trimethoxyphenyl)-9H-pyrido[3,4-b]indole (10)
2.3. Antimalarial Activity
2.3.1. Antiplasmodial Activity
2.3.2. In Vitro Cytotoxicity on Mammalian Cells
2.3.3. Selectivity Index (SI)
2.4. Evaluation of Physicochemical and Pharmacokinetic Properties
2.5. Evaluation of In Vivo Antimalarial Activity
2.5.1. Animals
2.5.2. Mouse Model of ECM
2.5.3. Drug Administration
2.5.4. Basic Indicator Evaluation
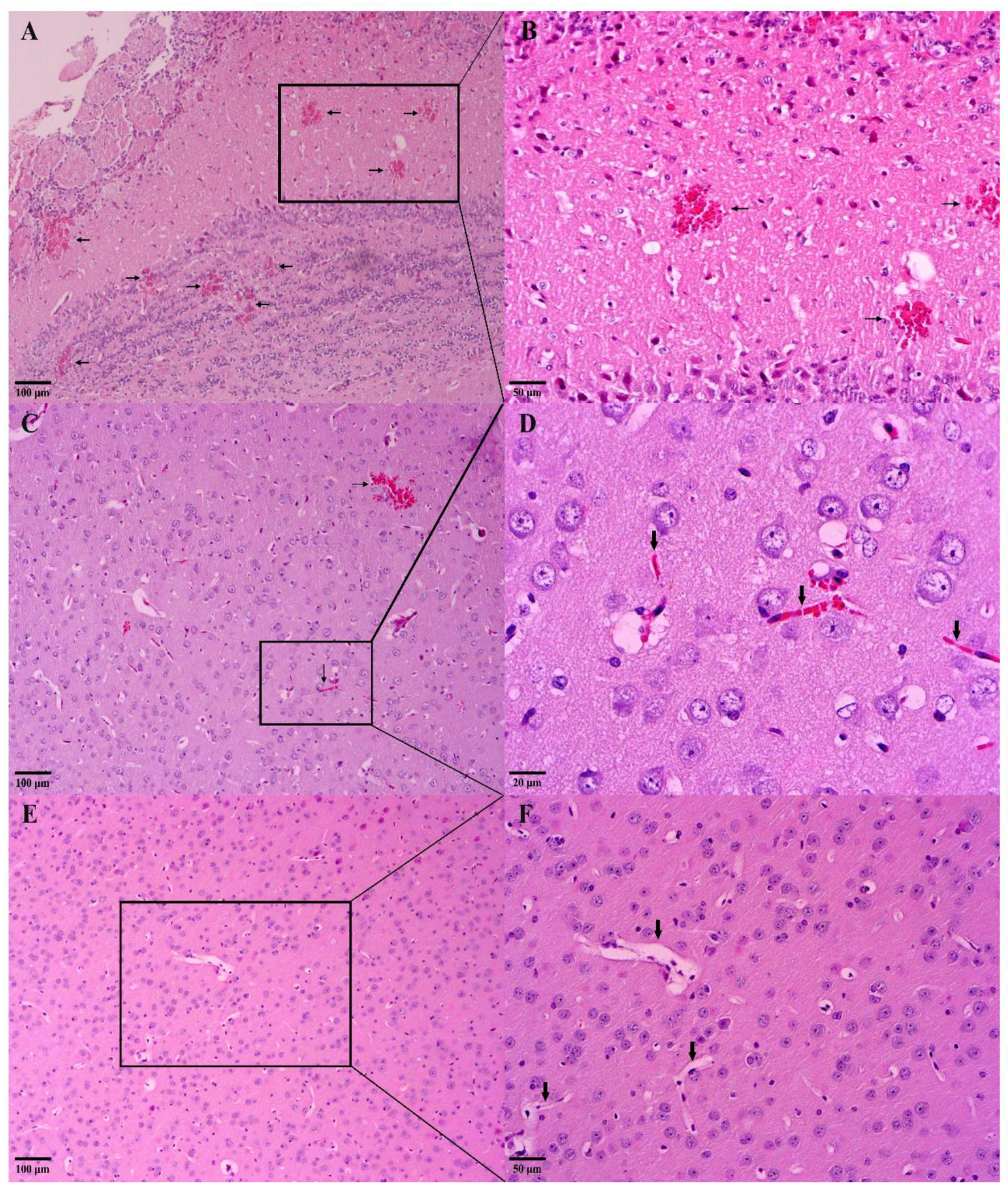
2.5.5. Macroscopic and Histological Analysis of the Brain
2.6. Evaluation of the In Silico and In Vitro Inhibition Potential of Nitric Oxide Synthesis
2.6.1. In Vitro Cytotoxicity against Mammalian Cells
2.6.2. Evaluation of Anti-Inflammatory Activity of Compound 10 by NO Dosage
2.6.3. Evaluation of the Interaction with iNOS by Molecular Docking Simulations
2.6.4. Statistical Analysis
3. Results and Discussion
3.1. Virtual Screening Results
3.2. Synthesis
3.3. Biological Evaluation
3.3.1. Cytotoxicity on Mammalian Cells and Antiplasmodial Activity
3.3.2. Evaluation of In Silico Physicochemical and Pharmacokinetic Parameters
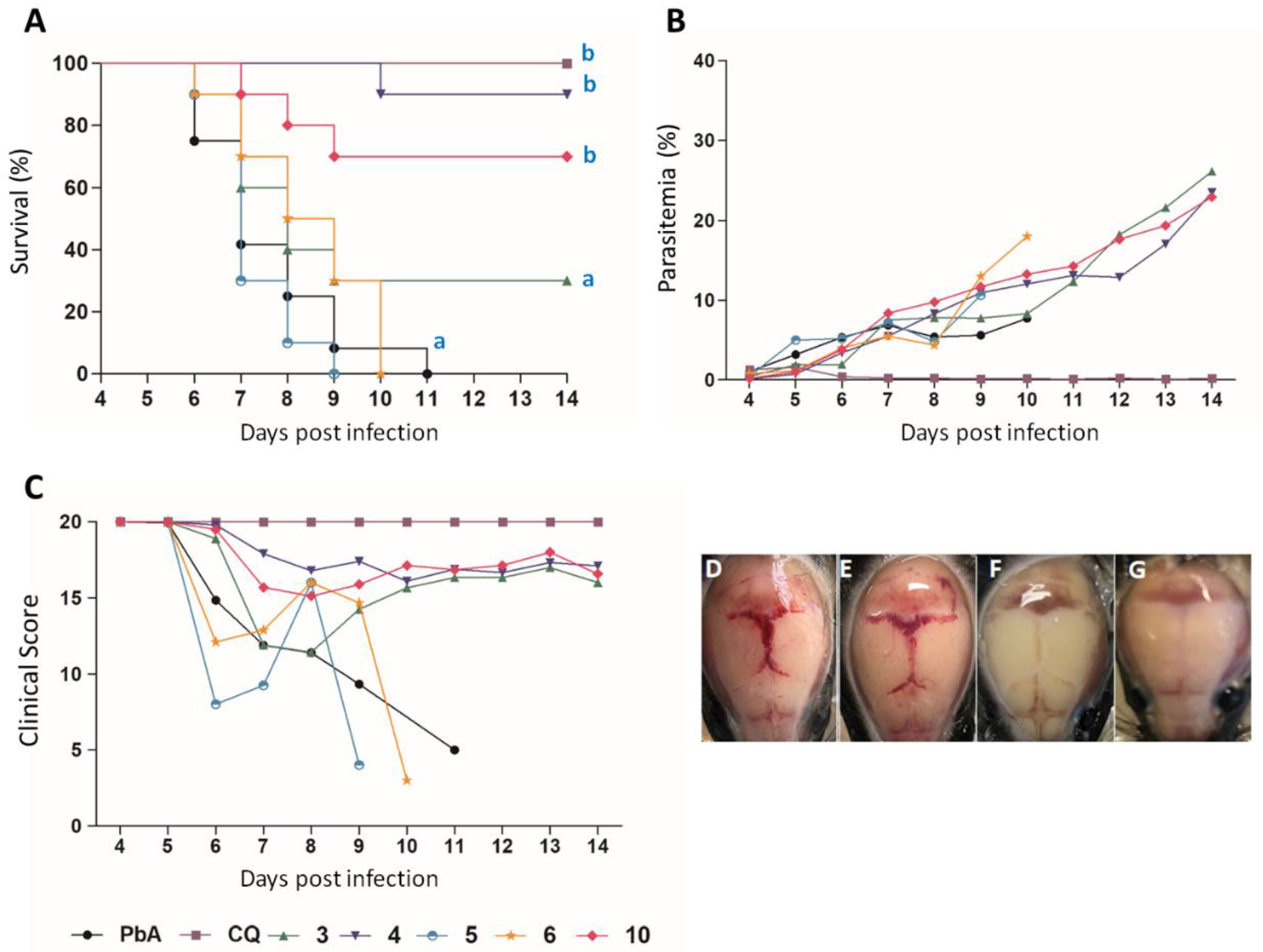
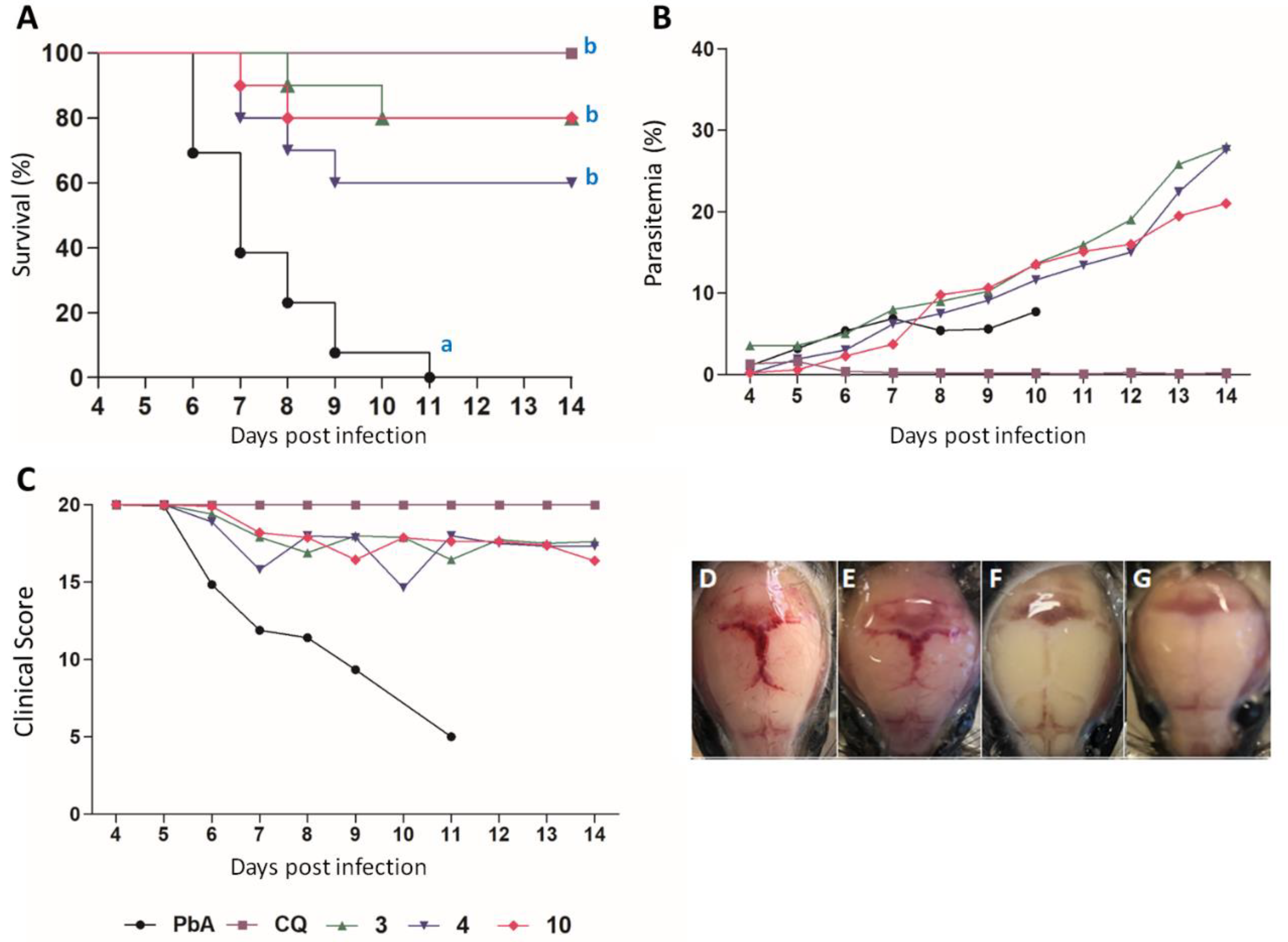
3.3.3. In Vivo Antimalarial Activity
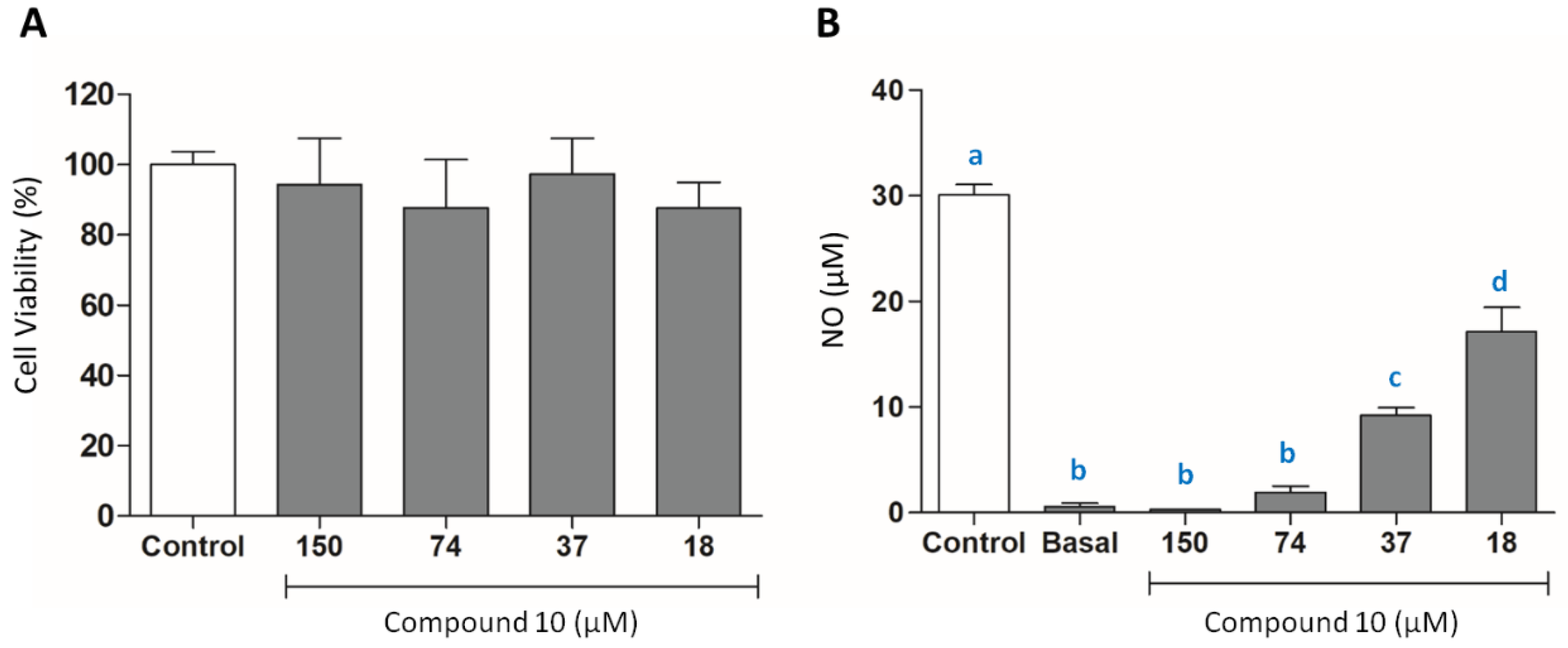
3.3.4. Evaluation of NO Production
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Guinovart, C.; Sigaúque, B.; Bassat, Q.; Loscertales, M.P.; Nhampossa, T.; Acácio, S.; Machevo, S.; Maculuve, S.; Bambo, G.; Mucavele, H.; et al. The Epidemiology of Severe Malaria at Manhiça District Hospital, Mozambique: A Retrospective Analysis of 20 Years of Malaria Admissions Surveillance Data. Lancet Glob. Health 2022, 10, e873–e881. [Google Scholar] [CrossRef]
- World Health Organization. WHO Guidelines for Malaria-31 March 2021; World Health Organization: Geneva, Switzerland, 2021; Volume 1, p. 210.
- Phillips, M.A.; Burrows, J.N.; Manyando, C.; Van Huijsduijnen, R.H.; Van Voorhis, W.C.; Wells, T.N.C. Malaria. Nat. Rev. Dis. Prim. 2017, 3, 17050. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, D.; Patnaik, J.K.; Mohanty, S.; Satpathy, S.K.; Das, B.S.; Mishra, S.K. Vascular Clogging, Mononuclear Cell Margination, and Enhanced Vascular Permeability in the Pathogenesis of Human Cerebral Malaria. Am. J. Trop. Med. Hyg. 1994, 51, 642–647. [Google Scholar] [CrossRef]
- Berendt, A.R.; Ferguson, D.J.P.; Gardner, J.; Turner, G.; Rowe, A.; McCormick, C.; Roberts, D.; Craig, A.; Pinches, R.; Elford, B.C.; et al. Molecular Mechanisms of Sequestration in Malaria. Parasitology 1994, 108, S19–S28. [Google Scholar] [CrossRef] [PubMed]
- Coban, C.; Lee, M.S.J.; Ishii, K.J. Tissue-Specific Immunopathology during Malaria Infection. Nat. Rev. Immunol. 2018, 18, 266–278. [Google Scholar] [CrossRef]
- Idro, R.; Marsh, K.; John, C.C.; Newton, C.R.J. Cerebral Malaria: Mechanisms of Brain Injury and Strategies for Improved Neurocognitive Outcome. Pediatr. Res. 2010, 68, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Boivin, M.J.; Bangirana, P.; Byarugaba, J.; Opoka, R.O.; Idro, R.; Jurek, A.M.; John, C.C. Cognitive Impairment After Cerebral Malaria in Children: A Prospective Study. Pediatrics 2007, 119, e360–e366. [Google Scholar] [CrossRef] [Green Version]
- Magen, J.; Taylor, T.; Brim, R.; Langfitt, J.; Mboma, S.; Semrud-Clikeman, M.; Kampondeni, S. Cognitive Outcomes and Psychiatric Symptoms of Retinopathy-Positive Cerebral Malaria: Cohort Description and Baseline Results. Am. J. Trop. Med. Hyg. 2017, 97, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Idro, R.; Jenkins, N.E.; Newton, C.R. Pathogenesis, Clinical Features, and Neurological Outcome of Cerebral Malaria. Lancet Neurol. 2005, 4, 827–840. [Google Scholar] [CrossRef]
- de Araújo, R.V.; Santos, S.S.; Sanches, L.M.; Giarolla, J.; El Seoud, O.; Ferreira, E.I. Malaria and Tuberculosis as Diseases of Neglected Populations: State of the Art in Chemotherapy and Advances in the Search for New Drugs. Mem. Inst. Oswaldo Cruz 2020, 115, e200229. [Google Scholar] [CrossRef] [PubMed]
- John, C.C.; Kutamba, E.; Mugarura, K.; Opoka, R.O. Adjunctive Therapy for Cerebral Malaria and Other Severe Forms of Plasmodium Falciparum Malaria. Expert Rev. Anti. Infect. Ther. 2010, 8, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Varo, R.; Erice, C.; Johnson, S.; Bassat, Q.; Kain, K.C. Clinical Trials to Assess Adjuvant Therapeutics for Severe Malaria. Malar. J. 2020, 19, 268. [Google Scholar] [CrossRef] [PubMed]
- Wicht, K.J.; Mok, S.; Fidock, D.A. Molecular Mechanisms of Drug Resistance in Plasmodium Falciparum Malaria. Annu. Rev. Microbiol. 2020, 74, 431–454. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Dan, W.; Schneider, U.; Wang, J. β-Carboline Alkaloid Monomers and Dimers: Occurrence, Structural Diversity, and Biological Activities. Eur. J. Med. Chem. 2018, 157, 622–656. [Google Scholar] [CrossRef] [PubMed]
- Jaromin, A.; Gryzło, B.; Jamrozik, M.; Parapini, S.; Basilico, N.; Cegła, M.; Taramelli, D.; Zagórska, A. Synthesis, Molecular Docking and Antiplasmodial Activities of New Tetrahydro-β-Carbolines. Int. J. Mol. Sci. 2021, 22, 13569. [Google Scholar] [CrossRef]
- Ashok, P.; Ganguly, S.; Murugesan, S. Review on In-Vitro Anti-Malarial Activity of Natural β-Carboline Alkaloids. Mini-Rev. Med. Chem. 2013, 13, 1778–1791. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Abraham, M.H.; Ibrahim, A.; Zissimos, A.M.; Zhao, Y.H.; Comer, J.; Reynolds, D.P. Application of Hydrogen Bonding Calculations in Property Based Drug Design. Drug Discov. Today 2002, 7, 1056–1063. [Google Scholar] [CrossRef]
- Gleeson, M.P. Generation of a Set of Simple, Interpretable ADMET Rules of Thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef]
- Leeson, P.D.; Davis, A.M.; Steele, J. Drug-like Properties: Guiding Principles for Design–or Chemical Prejudice? Drug Discov. Today Technol. 2004, 1, 189–195. [Google Scholar] [CrossRef]
- Wager, T.T.; Chandrasekaran, R.Y.; Hou, X.; Troutman, M.D.; Verhoest, P.R.; Villalobos, A.; Will, Y. Defining Desirable Central Nervous System Drug Space through the Alignment of Molecular Properties, in Vitro ADME, and Safety Attributes. ACS Chem. Neurosci. 2010, 1, 420–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.J.P. MOPAC: A Semiempirical Molecular Orbital Program. J. Comput. Aided. Mol. Des. 1990, 4, 1–103. [Google Scholar] [CrossRef]
- Dutra, J.D.L.; Filho, M.A.M.; Rocha, G.B.; Freire, R.O.; Simas, A.M.; Stewart, J.J.P. Sparkle/PM7 Lanthanide Parameters for the Modeling of Complexes and Materials. J. Chem. Theory Comput. 2013, 9, 3333–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doye, J.P.K.; Wales, D.J. Surveying a Potential Energy Surface by Eigenvector-Following. In Small Particles and Inorganic Clusters; Springer: Berlin/Heidelberg, Germany, 1997; Volume 197, pp. 194–197. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maia, E.H.B.; Campos, V.A.; dos Reis Santos, B.; Costa, M.S.; Lima, I.G.; Greco, S.J.; Ribeiro, R.I.M.A.; Munayer, F.M.; da Silva, A.M.; Taranto, A.G. Octopus: A Platform for the Virtual High-Throughput Screening of a Pool of Compounds against a Set of Molecular Targets. J. Mol. Model. 2017, 23, 26. [Google Scholar] [CrossRef]
- Nunes, R.R.; Fonseca, A.L.d.; Pinto, A.C.d.S.; Maia, E.H.B.; Silva, A.M.d.; Varotti, F.d.P.; Taranto, A.G. Brazilian Malaria Molecular Targets (BraMMT): Selected Receptors for Virtual High-Throughput Screening Experiments. Mem. Inst. Oswaldo Cruz 2019, 114, e180465. [Google Scholar] [CrossRef] [Green Version]
- Banfi, F.F.; Krombauer, G.C.; da Fonseca, A.L.; Nunes, R.R.; Andrade, S.N.; de Rezende, M.A.; Chaves, M.H.; dos Santos Monção Filho, E.; Taranto, A.G.; de Jesus Rodrigues, D.; et al. Dehydrobufotenin Extracted from the Amazonian Toad Rhinella Marina (Anura: Bufonidae) as a Prototype Molecule for the Development of Antiplasmodial Drugs. J. Venom. Anim. Toxins Incl. Trop. Dis. 2021, 27, e20200073. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium Falciparum Erythrocytic Stages in Culture. J. Parasitol. 1979, 65, 418. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J. Human Malaria Parasites in Continuous Culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Chiba, P.; Pferschy, S.; Vossen, M.G.; Noedl, H. The SYBR Green I Malaria Drug Sensitivity Assay: Performance in Low Parasitemia Samples. Am. J. Trop. Med. Hyg. 2010, 82, 398–401. [Google Scholar] [CrossRef]
- Costa Júnior, D.B.; Araújo, J.S.C.; de Mattos Oliveira, L.; Neri, F.S.M.; Moreira, P.O.L.; Taranto, A.G.; Fonseca, A.L.; de Pilla Varotti, F.; Leite, F.H.A. Identification of Novel Antiplasmodial Compound by Hierarquical Virtual Screening and in Vitro Assays. J. Biomol. Struct. Dyn. 2020, 39, 3378–3386. [Google Scholar] [CrossRef] [PubMed]
- Valsalam, S.; Agastian, P.; Esmail, G.A.; Ghilan, A.-K.M.; Al-Dhabi, N.A.; Arasu, M.V. Biosynthesis of Silver and Gold Nanoparticles Using Musa Acuminata Colla Flower and Its Pharmaceutical Activity against Bacteria and Anticancer Efficacy. J. Photochem. Photobiol. B Biol. 2019, 201, 111670. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.A.; Hall, J.E.; Kyle, D.E.; Grogl, M.; Ohemeng, K.A.; Allen, M.A.; Tidwell, R.R. Structure-Activity Relationships of Analogs of Pentamidine against Plasmodium Falciparum and Leishmania Mexicana Amazonensis. Antimicrob. Agents Chemother. 1990, 34, 1381–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Paramashivam, S.K.; Elayaperumal, K.; Natarajan, B.; Ramamoorthy, M.; Balasubramanian, S.; Dhiraviam, K. In Silico Pharmacokinetic and Molecular Docking Studies of Small Molecules Derived from Indigofera Aspalathoides Vahl Targeting Receptor Tyrosine Kinases. Bioinformation 2015, 11, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the General Solubility Equation: In Silico Prediction of Aqueous Solubility Incorporating the Effect of Topographical Polar Surface Area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef]
- National Research Council (US); Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press (US): Washington, DC, USA, 2011. [Google Scholar]
- Henning, A.N.; Roychoudhuri, R.; Restifo, N.P. Epigenetic Control of CD8+ T Cell Differentiation. Nat. Rev. Immunol. 2018, 18, 340–356. [Google Scholar] [CrossRef]
- Jiang, X.; Chen, L.; Zheng, Z.; Chen, Y.; Weng, X.; Guo, Y.; Li, K.; Yang, T.; Qu, S.; Liu, H.; et al. Synergistic Effect of Combined Artesunate and Tetramethylpyrazine in Experimental Cerebral Malaria. ACS Infect. Dis. 2020, 6, 2400–2409. [Google Scholar] [CrossRef]
- Medana, I.M.; Hunt, N.H.; Chan-Ling, T. Early Activation of Microglia in the Pathogenesis of Fatal Murine Cerebral Malaria. Glia 1997, 19, 91–103. [Google Scholar] [CrossRef]
- Carroll, R.W.; Wainwright, M.S.; Kim, K.Y.; Kidambi, T.; Gómez, N.D.; Taylor, T.; Haldar, K. A Rapid Murine Coma and Behavior Scale for Quantitative Assessment of Murine Cerebral Malaria. PLoS ONE 2010, 5, e13124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, W. Drug Resistance in Plasmodium Berghei. I. Chloroquine Resistance. Exp. Parasitol. 1965, 17, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Ngo-Thanh, H.; Sasaki, T.; Suzue, K.; Yokoo, H.; Isoda, K.; Kamitani, W.; Shimokawa, C.; Hisaeda, H.; Imai, T. Blood–Cerebrospinal Fluid Barrier: Another Site Disrupted during Experimental Cerebral Malaria Caused by Plasmodium Berghei ANKA. Int. J. Parasitol. 2020, 50, 1167–1175. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, X.; Broderick, M.; Fein, H. Measurement of Nitric Oxide Production in Biological Systems by Using Griess Reaction Assay. Sensors 2003, 3, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Xia, G.-Y.; Yao, T.; Zhang, B.-Y.; Li, Y.; Kang, N.; Cao, S.-J.; Ding, L.-Q.; Chen, L.-X.; Qiu, F. Withapubesides A–D: Natural Inducible Nitric Oxide Synthase (INOS) Inhibitors from Physalis Pubescens. Org. Biomol. Chem. 2017, 15, 10016–10023. [Google Scholar] [CrossRef]
- Milani, M.; Balconi, E.; Aliverti, A.; Mastrangelo, E.; Seeber, F.; Bolognesi, M.; Zanetti, G. Ferredoxin-NADP+ Reductase from Plasmodium Falciparum Undergoes NADP+-Dependent Dimerization and Inactivation: Functional and Crystallographic Analysis. J. Mol. Biol. 2007, 367, 501–513. [Google Scholar] [CrossRef]
- Balconi, E.; Pennati, A.; Crobu, D.; Pandini, V.; Cerutti, R.; Zanetti, G.; Aliverti, A. The Ferredoxin-NADP + Reductase/Ferredoxin Electron Transfer System of Plasmodium Falciparum. FEBS J. 2009, 276, 3825–3836. [Google Scholar] [CrossRef]
- Lesanavičius, M.; Aliverti, A.; Šarlauskas, J.; Čėnas, N. Reactions of Plasmodium Falciparum Ferredoxin:NADP+ Oxidoreductase with Redox Cycling Xenobiotics: A Mechanistic Study. Int. J. Mol. Sci. 2020, 21, 3234. [Google Scholar] [CrossRef]
- Brokamp, R.; Bergmann, B.; Müller, I.B.; Bienz, S. Stereoselective Preparation of Pyridoxal 1,2,3,4-Tetrahydro-β-Carboline Derivatives and the Influence of Their Absolute and Relative Configuration on the Proliferation of the Malaria Parasite Plasmodium Falciparum. Bioorg. Med. Chem. 2014, 22, 1832–1837. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Ghavami, M.; Butler, J.H.; Merino, E.F.; Slebodnick, C.; Cassera, M.B.; Carlier, P.R. Probing the B- & C-Rings of the Antimalarial Tetrahydro-β-Carboline MMV008138 for Steric and Conformational Constraints. Bioorg. Med. Chem. Lett. 2020, 30, 127520. [Google Scholar] [CrossRef] [PubMed]
- Almolhim, H.; Ding, S.; Butler, J.H.; Bremers, E.K.; Butschek, G.J.; Slebodnick, C.; Merino, E.F.; Rizopoulos, Z.; Totrov, M.; Cassera, M.B.; et al. Enantiopure Benzofuran-2-Carboxamides of 1-Aryltetrahydro-β-Carbolines Are Potent Antimalarials In Vitro. ACS Med. Chem. Lett. 2022, 13, 371–376. [Google Scholar] [CrossRef]
- Mathew, J.; Ding, S.; Kunz, K.A.; Stacy, E.E.; Butler, J.H.; Haney, R.S.; Merino, E.F.; Butschek, G.J.; Rizopoulos, Z.; Totrov, M.; et al. Malaria Box-Inspired Discovery of N -Aminoalkyl-β-Carboline-3-Carboxamides, a Novel Orally Active Class of Antimalarials. ACS Med. Chem. Lett. 2022, 13, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Merckx, A.; Echalier, A.; Langford, K.; Sicard, A.; Langsley, G.; Joore, J.; Doerig, C.; Noble, M.; Endicott, J. Structures of P. Falciparum Protein Kinase 7 Identify an Activation Motif and Leads for Inhibitor Design. Structure 2008, 16, 228–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahlfs, S.; Fischer, M.; Becker, K. Plasmodium Falciparum Possesses a Classical Glutaredoxin and a Second, Glutaredoxin-like Protein with a PICOT Homology Domain. J. Biol. Chem. 2001, 276, 37133–37140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, S.; Pulcini, S.; Fatih, F.; Staines, H. Artemisinins and the Biological Basis for the PfATP6/SERCA Hypothesis. Trends Parasitol. 2010, 26, 517–523. [Google Scholar] [CrossRef]
- Yao, Z.-K.; Krai, P.M.; Merino, E.F.; Simpson, M.E.; Slebodnick, C.; Cassera, M.B.; Carlier, P.R. Determination of the Active Stereoisomer of the MEP Pathway-Targeting Antimalarial Agent MMV008138, and Initial Structure–Activity Studies. Bioorg. Med. Chem. Lett. 2015, 25, 1515–1519. [Google Scholar] [CrossRef] [Green Version]
- Poje, G.; Pessanha de Carvalho, L.; Held, J.; Moita, D.; Prudêncio, M.; Perković, I.; Tandarić, T.; Vianello, R.; Rajić, Z. Design and Synthesis of Harmiquins, Harmine and Chloroquine Hybrids as Potent Antiplasmodial Agents. Eur. J. Med. Chem. 2022, 238, 114408. [Google Scholar] [CrossRef]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; Van Huijsduijnen, R.H.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and Lead Criteria in Drug Discovery for Infectious Diseases of the Developing World. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef]
- Lin, J.; Sahakian, D.; de Morais, S.; Xu, J.; Polzer, R.; Winter, S. The Role of Absorption, Distribution, Metabolism, Excretion and Toxicity in Drug Discovery. Curr. Top. Med. Chem. 2003, 3, 1125–1154. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.E.; Scioli Montoto, S. Routes of Drug Administration. In ADME Processes in Pharmaceutical Sciences; Springer International Publishing: Cham, Switzerland, 2018; pp. 97–133. [Google Scholar]
- He, X.; Yan, J.; Zhu, X.; Wang, Q.; Pang, W.; Qi, Z.; Wang, M.; Luo, E.; Parker, D.M.; Cantorna, M.T.; et al. Vitamin D Inhibits the Occurrence of Experimental Cerebral Malaria in Mice by Suppressing the Host Inflammatory Response. J. Immunol. 2014, 193, 1314–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewick, P. Medicinal Natural Products: A Biosynthetic Aprproach, 2nd ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2002; ISBN 0471496413. [Google Scholar]
- Ryan, K.S.; Drennan, C.L. Divergent Pathways in the Biosynthesis of Bisindole Natural Products. Chem. Biol. 2009, 16, 351–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talisuna, A.O.; Bloland, P.; D’Alessandro, U. History, Dynamics, and Public Health Importance of Malaria Parasite Resistance. Clin. Microbiol. Rev. 2004, 17, 235–254. [Google Scholar] [CrossRef] [Green Version]
- Achan, J.; Talisuna, A.O.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an Old Anti-Malarial Drug in a Modern World: Role in the Treatment of Malaria. Malar. J. 2011, 10, 144. [Google Scholar] [CrossRef] [Green Version]
- Andrade-Neto, V.F.; Brandão, M.G.L.; Stehmann, J.R.; Oliveira, L.A.; Krettli, A.U. Antimalarial Activity of Cinchona-like Plants Used to Treat Fever and Malaria in Brazil. J. Ethnopharmacol. 2003, 87, 253–256. [Google Scholar] [CrossRef]
- Fiot, J.; Sanon, S.; Azas, N.; Mahiou, V.; Jansen, O.; Angenot, L.; Balansard, G.; Ollivier, E. Phytochemical and Pharmacological Study of Roots and Leaves of Guiera Senegalensis J.F. Gmel (Combretaceae). J. Ethnopharmacol. 2006, 106, 173–178. [Google Scholar] [CrossRef]
- Takasu, K.; Shimogama, T.; Saiin, C.; Kim, H.-S.; Wataya, Y.; Brun, R.; Ihara, M. Synthesis and Evaluation of β-Carbolinium Cations as New Antimalarial Agents Based on π-Delocalized Lipophilic Cation (DLC) Hypothesis. Chem. Pharm. Bull. 2005, 53, 653–661. [Google Scholar] [CrossRef] [Green Version]
- Yenjai, C.; Sripontan, S.; Sriprajun, P.; Kittakoop, P.; Jintasirikul, A.; Tanticharoen, M.; Thebtaranonth, Y. Coumarins and Carbazoles with Antiplasmodial Activity from Clausena Harmandiana. Planta Med. 2000, 66, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Gorki, V.; Walter, N.S.; Singh, R.; Chauhan, M.; Dhingra, N.; Salunke, D.B.; Kaur, S. β-Carboline Derivatives Tackling Malaria: Biological Evaluation and Docking Analysis. ACS Omega 2020, 5, 17993–18006. [Google Scholar] [CrossRef]
- Wassmer, S.C.; Emile, G.; Grau, R. Severe Malaria: What’s New on the Pathogenesis Front? Graphical Abstract HHS Public Access. Int. J. Parasitol 2017, 47, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Sierro, F.; Grau, G.E.R. The Ins and Outs of Cerebral Malaria Pathogenesis: Immunopathology, Extracellular Vesicles, Immunometabolism, and Trained Immunity. Front. Immunol. 2019, 10, 830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazan, M.; Hdayati, M. The Role of Nitric Oxide in Health and Diseases. Scimetr 2014, 4, 38–43. [Google Scholar] [CrossRef]
- Brown, G.C. Nitric Oxide and Neuronal Death. Nitric Oxide 2010, 23, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Brunet, L.R. Nitric Oxide in Parasitic Infections. Int. Immunopharmacol. 2001, 1, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.S.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric Oxide and Redox Mechanisms in the Immune Response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and Stroke: Identifying Novel Targets for Neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [Green Version]
- Fujikawa, D.G. The Role of Excitotoxic Programmed Necrosis in Acute Brain Injury. Comput. Struct. Biotechnol. J. 2015, 13, 212–221. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Wolan, D.; Adak, S.; Crane, B.R.; Kwon, N.S.; Tainer, J.A.; Getzoff, E.D.; Stuehr, D.J. Mutational Analysis of the Tetrahydrobiopterin-Binding Site in Inducible Nitric-Oxide Synthase. J. Biol. Chem. 1999, 274, 24100–24112. [Google Scholar] [CrossRef] [Green Version]
- Crane, B.R.; Arvai, A.S.; Ghosh, D.K.; Wu, C.; Getzoff, E.D.; Stuehr, D.J.; Tainer, J.A. Structure of Nitric Oxide Synthase Oxygenase Dimer with Pterin and Substrate. Science 1998, 279, 2121–2126. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | ||||||||
|---|---|---|---|---|---|---|---|---|
| Models | Crystallographic Ligand | 3 | 4 | 5 | 6 | 8 | 9 | 10 |
| 2OK8 | −2.0 | −5.3 | −5.0 | −5.2 | −5.0 | −4.9 | −4.8 | −4.7 |
| 2PML | −6.9 | −9.0 | −8.8 | −9.4 | −9.0 | −8.6 | −7.9 | −8.8 |
| 4N0Z | −4.3 | −5.8 | −6.1 | −6.1 | −6.3 | −5.6 | −5.3 | −5.5 |
| PfATP6 | −7.2 | −8.9 | −8.3 | −9.0 | −8.8 | −7.8 | −7.4 | −7.4 |
| Compounds | P. falciparum IC50 ± SD (μM) * | WI-26-VA4 IC50 ± SD (μM) * | SI |
|---|---|---|---|
| 3 | 0.71 ± 0.012 | >100 | >141 |
| 4 | 1.04 ± 0.021 | >100 | >96 |
| 5 | 1.41 ± 0.018 | >100 | >71 |
| 6 | 0.67 ± 0.019 | >100 | >149 |
| 8 | 1.82 ± 0.011 | >100 | >55 |
| 9 | 1.12 ± 0.014 | >100 | >89 |
| 10 | 0.51 ± 0.011 | >100 | >196 |
| ART ** | 0.095 ±0.010 | >100 | >1000 |
| CQ *** | 0.59 ± 0.015 | >100 | >169 |
| Compound | HIE (p) | Caco-2 (p) | BBB (p) |
|---|---|---|---|
| 3 | +(0.98) | +(0.84) | +(0.95) |
| 4 | +(0.98) | +(0.84) | +(0.95) |
| 5 | +(0.98) | +(0.87) | +(0.97) |
| 6 | +(0.98) | +(0.87) | +(0.97) |
| 10 | +(0.99) | +(0.82) | +(0.96) |
| CQ | +(0.99) | +(0.66) | +(1.00) |
| Compound | Dosage (mg/Kg) | Parasitemia ± SD (% Reduction) | n | |
|---|---|---|---|---|
| 5th dpi | 7th dpi | |||
| 3 | 10 | 1.87 ± 1.02 b (41.19) | 7.50 ± 2.49 (0) | 10 |
| 4 | 10 | 0.79 ± 0.63 a,b (75.16) | 5.48± 3.64 (20.00) | 10 |
| 5 | 10 | 5.00 ± 1.59 (0.00) | 7.20 ± 4.57 (0) | 10 |
| 6 | 10 | 1.13 ± 0.48 a,b (64.36) | 5.48 ± 3.27 (0) | 10 |
| 10 | 10 | 1.02 ± 0.93 a,b (67.92) | 8.37 ± 2.26 (0) | 10 |
| CQ * | 10 | 0.24 ± 0.05 a (96.5) | 0.21 ± 0.27 a (96.1) | 09 |
| Water | - | 3.18 ± 2.68 | 6.85 ± 2.57 | 13 |
| Compound | Dosage (mg/Kg) | Parasitemia ± SD (% Reduction) | n | |
|---|---|---|---|---|
| 5th dpi | 7th dpi | |||
| 3 | 10 | 3.56 ± 2.57 (0.00) | 7.96 ± 3.26 (0) | 10 |
| 4 | 10 | 1.88 ± 2.46 b (40.88) | 6.18 ± 4.75 (9.78) | 10 |
| 10 | 10 | 0.57 ± 0.94 a,b (82.07) | 3.72 ± 3.93 b (45.69) | 10 |
| CQ * | 10 | 0.24 ± 0.05 a (96.5) | 0.21 ± 0.27 a (96.1) | 9 |
| Water | - | 3.18 ± 2.68 | 6.85 ± 2.57 | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, F.d.M.; Bellei, J.C.B.; Barbosa, C.d.S.; Duarte, C.L.; Fonseca, A.L.d.; Pinto, A.C.d.S.; Raimundo, F.O.; Carpinter, B.A.; Lemos, A.S.d.O.; Coimbra, E.S.; et al. Rational-Based Discovery of Novel β-Carboline Derivatives as Potential Antimalarials: From In Silico Identification of Novel Targets to Inhibition of Experimental Cerebral Malaria. Pathogens 2022, 11, 1529. https://doi.org/10.3390/pathogens11121529
Alves FdM, Bellei JCB, Barbosa CdS, Duarte CL, Fonseca ALd, Pinto ACdS, Raimundo FO, Carpinter BA, Lemos ASdO, Coimbra ES, et al. Rational-Based Discovery of Novel β-Carboline Derivatives as Potential Antimalarials: From In Silico Identification of Novel Targets to Inhibition of Experimental Cerebral Malaria. Pathogens. 2022; 11(12):1529. https://doi.org/10.3390/pathogens11121529
Chicago/Turabian StyleAlves, Fernanda de Moura, Jessica Correa Bezerra Bellei, Camila de Souza Barbosa, Caíque Lopes Duarte, Amanda Luisa da Fonseca, Ana Claudia de Souza Pinto, Felipe Oliveira Raimundo, Bárbara Albuquerque Carpinter, Ari Sérgio de Oliveira Lemos, Elaine Soares Coimbra, and et al. 2022. "Rational-Based Discovery of Novel β-Carboline Derivatives as Potential Antimalarials: From In Silico Identification of Novel Targets to Inhibition of Experimental Cerebral Malaria" Pathogens 11, no. 12: 1529. https://doi.org/10.3390/pathogens11121529