
Akt Inhibition Promotes Autophagy and Clearance of Group B Streptococcus from the Alveolar Epithelium
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
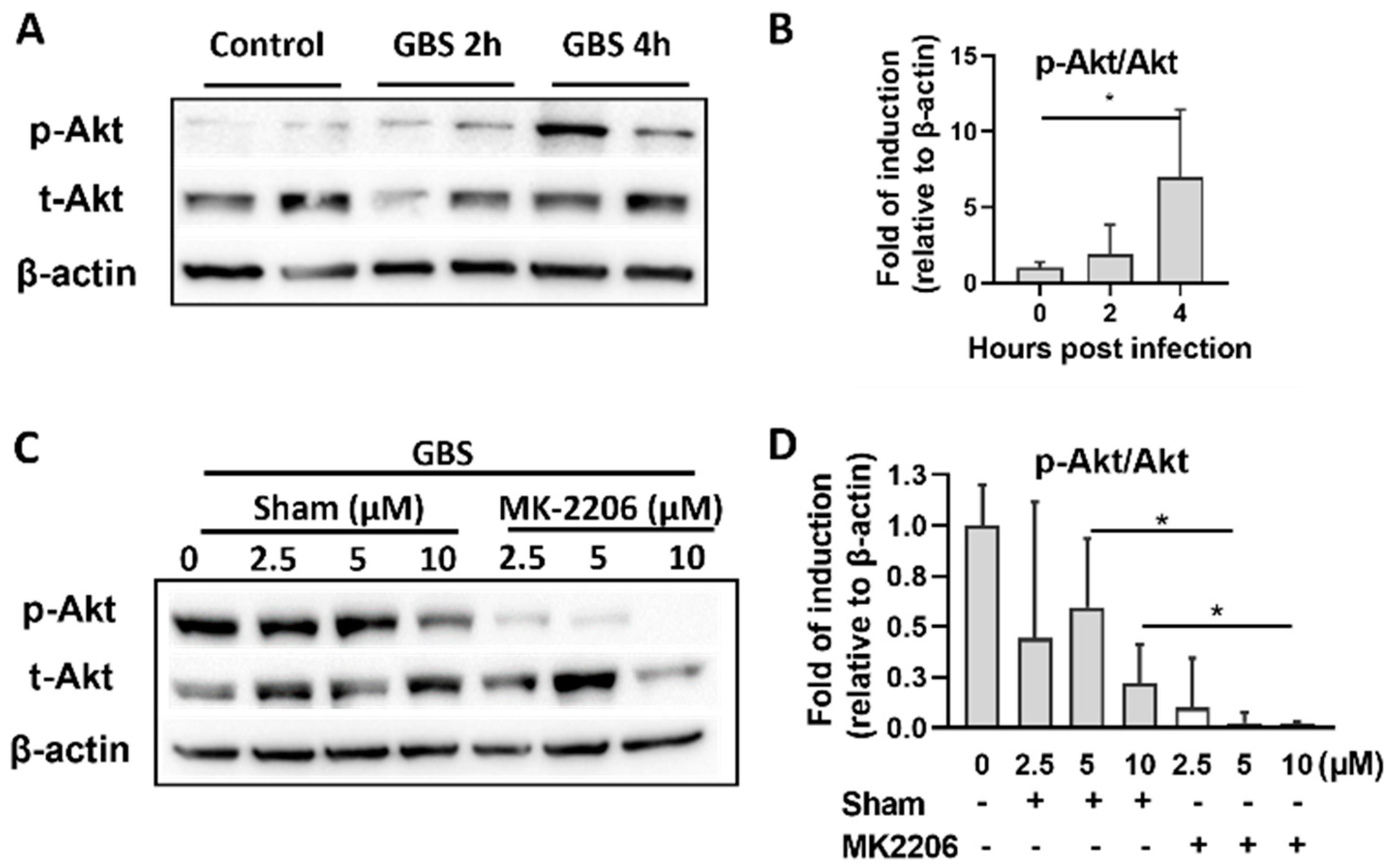
2.1. GBS Infection Activates Akt in Alveolar Epithelial Cells
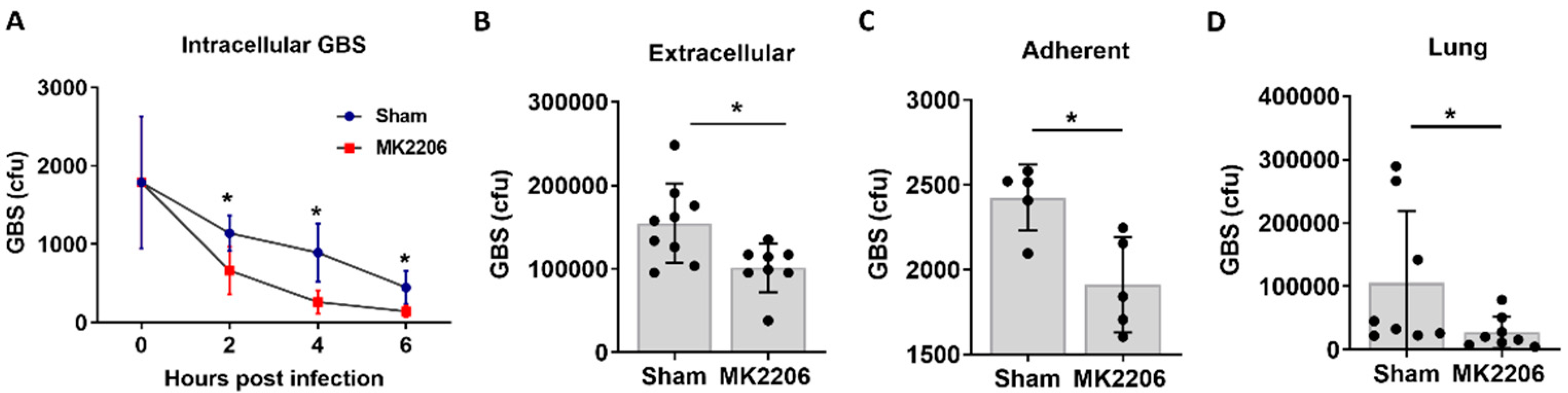
2.2. Inhibition of Akt Kinase Promotes GBS Clearance by Lung Epithelial Cells
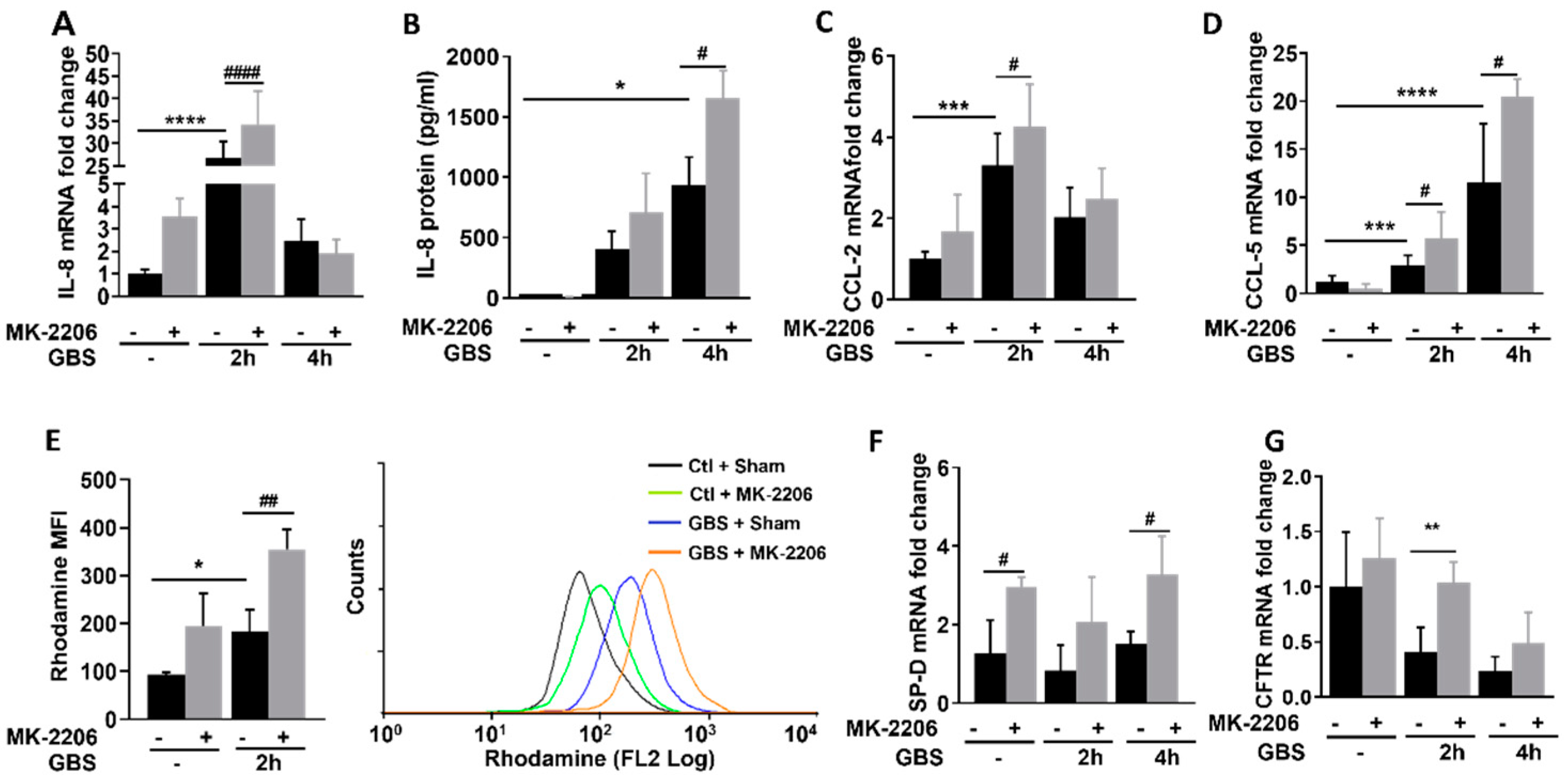
2.3. Akt Inhibition Promotes Inflammatory Responses in GBS Infected Epithelial Cells
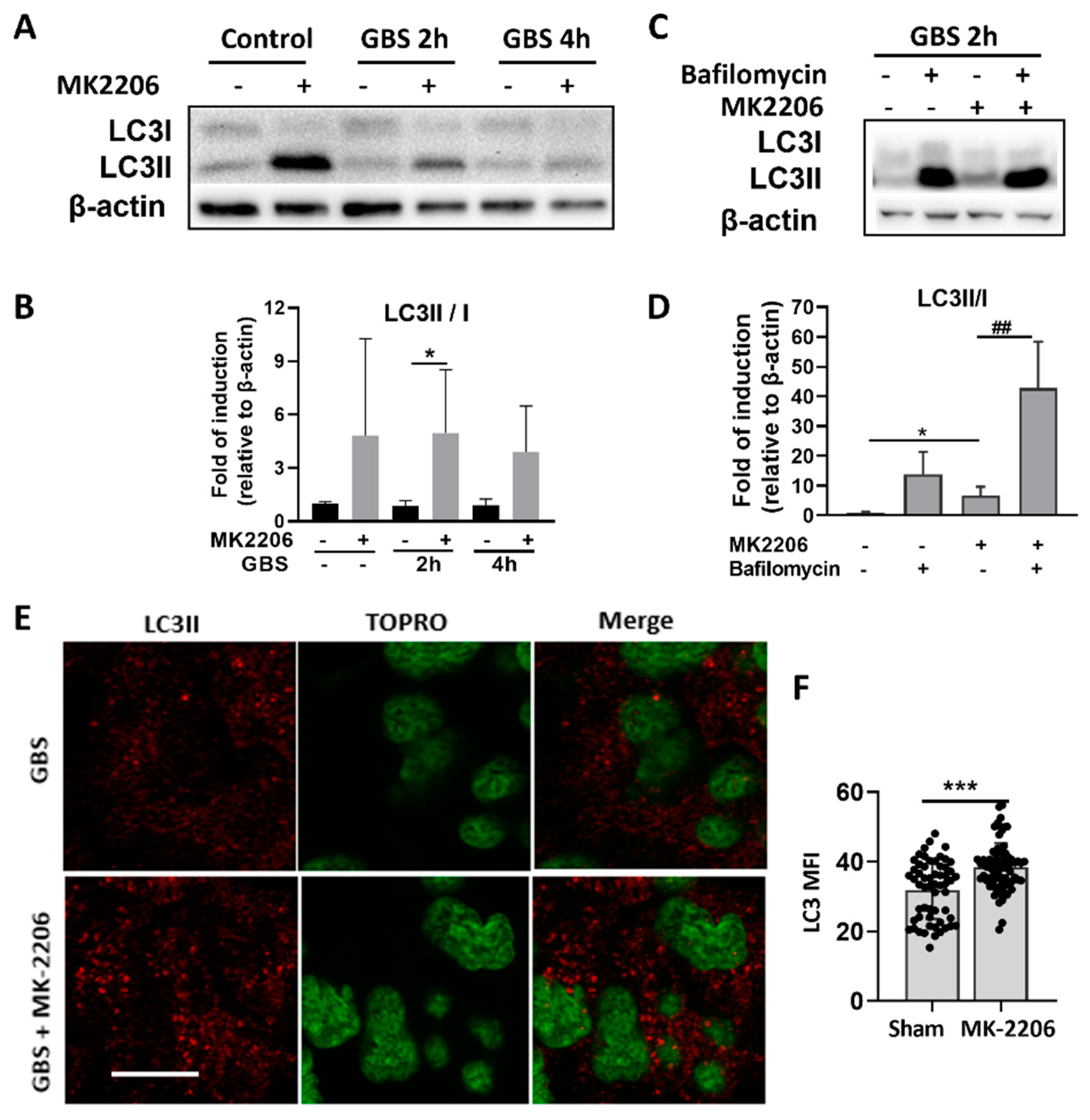
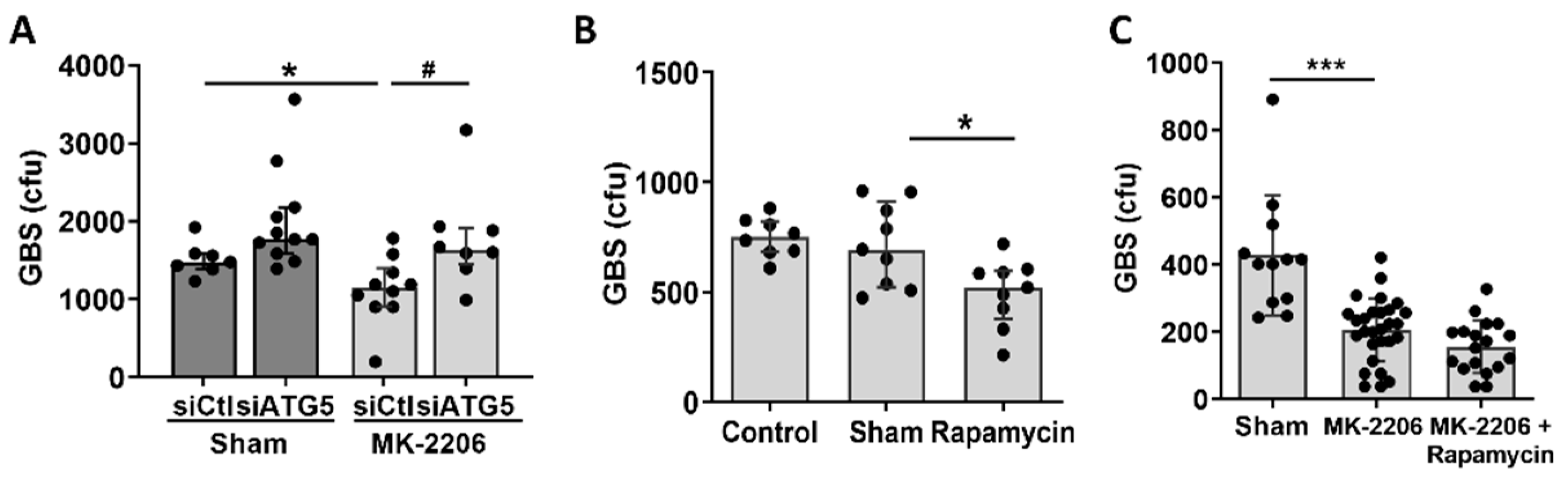
2.4. Akt Inhibition Induces GBS Clearance in Alveolar Epithelial Cells via Induction of Autophagy
3. Discussion
4. Materials and Methods
4.1. GBS Strain and Culture
4.2. GBS Survival Assay in Alveolar Epithelial Cells
4.3. Murine Model of GBS Pneumonia
4.4. In Vivo Murine Sample Collection, Analysis, and Survival Experiments
4.5. Cell Proliferation Assay
4.6. RNA Isolation and Real-Time PCR
4.7. Enzyme-Linked Immunoabsorbent Assay (ELISA)
4.8. Western Blot
4.9. Gene Silencing
4.10. Measurement of Reactive Oxygen Species
4.11. Immunofluorescence Staining
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shet, A.; Ferrieri, P. Neonatal & maternal group B streptococcal infections: A comprehensive review. Indian J. Med. Res. 2004, 120, 141–150. [Google Scholar] [PubMed]
- Barcaite, E.; Bartusevicius, A.; Tameliene, R.; Kliucinskas, M.; Maleckiene, L.; Nadisauskiene, R. Prevalence of maternal group B streptococcal colonisation in European countries. Acta Obstet. Gynecol. Scand. 2008, 87, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Vergadi, E.; Manoura, A.; Chatzakis, E.; Karavitakis, E.; Maraki, S.; Galanakis, E. Changes in the incidence and epidemiology of neonatal group B Streptococcal disease over the last two decades in Crete, Greece. Infect. Dis. Rep. 2018, 10, 7744. [Google Scholar] [CrossRef]
- Rubens, C.E.; Smith, S.; Hulse, M.; Chi, E.Y.; Van Belle, G. Respiratory epithelial cell invasion by group B streptococci. Infect. Immun. 1992, 60, 5157–5163. [Google Scholar] [CrossRef]
- Valentin-Weigand, P.; Chhatwal, G.S. Correlation of epithelial cell invasiveness of group B streptococci with clinical source of isolation. Microb. Pathog. 1995, 19, 83–91. [Google Scholar] [CrossRef]
- Eddens, T.; Kolls, J.K. Host defenses against bacterial lower respiratory tract infection. Curr. Opin. Immunol. 2012, 24, 424–430. [Google Scholar] [CrossRef]
- Nizet, V.; Kim, K.S.; Stins, M.; Jonas, M.; Chi, E.Y.; Nguyen, D.; E Rubens, C. Invasion of brain microvascular endothelial cells by group B streptococci. Infect. Immun. 1997, 65, 5074–5081. [Google Scholar] [CrossRef]
- Cutting, A.S.; Del Rosario, Y.; Mu, R.; Rodriguez, A.; Till, A.; Subramani, S.; Gottlieb, R.A.; Doran, K.S. The role of autophagy during group B Streptococcus infection of blood-brain barrier endothelium. J. Biol. Chem. 2014, 289, 35711–35723. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef]
- Knodler, L.A.; Celli, J. Eating the strangers within: Host control of intracellular bacteria via xenophagy. Cell. Microbiol. 2011, 13, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, I.; Amano, A.; Mizushima, N.; Yamamoto, A.; Yamaguchi, H.; Kamimoto, T.; Nara, A.; Funao, J.; Nakata, M.; Tsuda, K.; et al. Autophagy defends cells against invading group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- O’Keeffe, K.M.; Wilk, M.M.; Leech, J.M.; Murphy, A.G.; Laabei, M.; Monk, I.R.; Massey, R.C.; Lindsay, J.; Foster, T.J.; Geoghegan, J.A.; et al. Manipulation of Autophagy in Phagocytes Facilitates Staphylococcus aureus Bloodstream Infection. Infect. Immun. 2015, 83, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagy in infection. Curr. Opin. Cell Biol. 2010, 22, 252–262. [Google Scholar] [CrossRef]
- Martinez, J.; Malireddi, R.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.-L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef]
- Gong, L.; Devenish, R.J.; Prescott, M. Autophagy as a macrophage response to bacterial infection. IUBMB Life 2012, 64, 740–747. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Androulidaki, A.; Iliopoulos, D.; Arranz, A.; Doxaki, C.; Schworer, S.; Zacharioudaki, V.; Margioris, A.N.; Tsichlis, P.N.; Tsatsanis, C. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity 2009, 31, 220–231. [Google Scholar] [CrossRef]
- Kuijl, C.; Savage, N.D.L.; Marsman, M.; Tuin, A.W.; Janssen, L.; Egan, D.A.; Ketema, M.; Van den Nieuwendijk, R.; Van den Eeden, S.J.; Geluk, A.; et al. Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature 2007, 450, 725–730. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Kolliniati, O.; Ieronymaki, E.; Vergadi, E.; Tsatsanis, C. Metabolic Regulation of Macrophage Activation. J. Innate Immun. 2021, 14, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, T. Autophagy vs. Group A Streptococcus. Autophagy 2006, 2, 154–155. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Bi, Y.; Wang, R.; Shen, B.; Zhang, Y.; Yang, H.; Wang, X.; Liu, H.; Lu, Y.; Han, F. Kinase AKT1 negatively controls neutrophil recruitment and function in mice. J. Immunol. 2013, 191, 2680–2690. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Baltimore, R.S.; Huie, S.M.; Meek, J.I.; Schuchat, A.; O’Brien, K.L. Early-onset neonatal sepsis in the era of group B streptococcal prevention. Pediatrics 2001, 108, 1094–1098. [Google Scholar] [CrossRef]
- Lambert, L.; Culley, F.J. Innate Immunity to Respiratory Infection in Early Life. Front. Immunol. 2017, 8, 1570. [Google Scholar] [CrossRef]
- Iida, M.; Brand, T.M.; Campbell, D.A.; Starr, M.M.; Luthar, N.; Traynor, A.M.; Wheeler, D.L. Targeting AKT with the allosteric AKT inhibitor MK-2206 in non-small cell lung cancer cells with acquired resistance to cetuximab. Cancer Biol. Ther. 2013, 14, 481–491. [Google Scholar] [CrossRef]
- Burnham, C.D.; Shokoples, S.E.; Tyrrell, G.J. Invasion of HeLa cells by group B streptococcus requires the phosphoinositide-3-kinase signalling pathway and modulates phosphorylation of host-cell Akt and glycogen synthase kinase-3. Microbiology 2007, 153 Pt 12, 4240–4252. [Google Scholar] [CrossRef]
- Owen, K.A.; Meyer, C.B.; Bouton, A.H.; Casanova, J.E. Activation of focal adhesion kinase by Salmonella suppresses autophagy via an Akt/mTOR signaling pathway and promotes bacterial survival in macrophages. PLoS Pathog. 2014, 10, e1004159. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, G.; Midiri, A.; Beninati, C.; Biondo, C.; Galbo, R.; Akira, S.; Henneke, P.; Golenbock, D.; Teti, G. Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J. Immunol. 2004, 172, 6324–6329. [Google Scholar] [CrossRef] [PubMed]
- Nandan, D.; Zhang, N.; Yu, Y.; Schwartz, B.; Chen, S.; Kima, P.E.; Reiner, N.E. Miransertib (ARQ 092), an orally-available, selective Akt inhibitor is effective against Leishmania. PLoS ONE. 2018, 13, e0206920. [Google Scholar] [CrossRef] [PubMed]
- Bals, R.; Hiemstra, P.S. Innate immunity in the lung: How epithelial cells fight against respiratory pathogens. Eur. Respir. J. 2004, 23, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Mikamo, H.; Johri, A.K.; Paoletti, L.C.; Madoff, L.C.; Onderdonk, A.B. Adherence to, invasion by, and cytokine production in response to serotype VIII group B Streptococci. Infect. Immun. 2004, 72, 4716–4722. [Google Scholar] [CrossRef]
- Eisele, N.A.; Anderson, D.M. Host Defense and the Airway Epithelium: Frontline Responses That Protect against Bacterial Invasion and Pneumonia. J. Pathog. 2011, 2011, 249802. [Google Scholar] [CrossRef]
- Paraskakis, E.; Vergadi, E.; Chatzimichael, A.; Bush, A. The Role of Flow-Independent Exhaled Nitric Oxide Parameters in the Assessment of Airway Diseases. Curr. Top. Med. Chem. 2016, 16, 1631–1642. [Google Scholar] [CrossRef]
- Leiva-Juarez, M.M.; Kolls, J.K.; Evans, S.E. Lung epithelial cells: Therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. 2018, 11, 21–34. [Google Scholar] [CrossRef]
- Soriani, M.; Santi, I.; Taddei, A.; Rappuoli, R.; Grandi, G.; Telford, J.L. Group B Streptococcus crosses human epithelial cells by a paracellular route. J. Infect. Dis. 2006, 193, 241–250. [Google Scholar] [CrossRef]
- Haspel, J.A.; Choi, A.M. Autophagy: A core cellular process with emerging links to pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 1237–1246. [Google Scholar] [CrossRef]
- Yuan, K.; Huang, C.; Fox, J.; Laturnus, D.; Carlson, E.; Zhang, B.; Yin, Q.; Gao, H.; Wu, M. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J. Cell Sci. 2012, 125, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.G.; Ji, T.X.; Xia, Y.; Ma, Y.Y. Autophagy protects type II alveolar epithelial cells from Mycobacterium tuberculosis infection. Biochem. Biophys. Res. Commun. 2013, 432, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009, 5, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Radtke, A.L.; O’Riordan, M.X. Intracellular innate resistance to bacterial pathogens. Cell Microbiol. 2006, 8, 1720–1729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, X.; Kang, J.; Su, J.; Li, Y.; Zhong, J.; Sun, L. Oxidative stress induces parallel autophagy and mitochondria dysfunction in human glioma U251 cells. Toxicol. Sci. 2009, 110, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Farrell, F.O.; Rusten, T.E.; Stenmark, H. Phosphoinositide 3-kinases as accelerators and brakes of autophagy. FEBS J. 2013, 280, 63237. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef]
- Mitchell, A.M.; Mitchell, T.J. Streptococcus pneumoniae: Virulence factors and variation. Clin. Microbiol. Infect. 2010, 16, 411–418. [Google Scholar] [CrossRef]
- Li, P.; Shi, J.; He, Q.; Hu, Q.; Wang, Y.Y.; Zhang, L.J.; Chan, W.T.; Chen, W. Streptococcus pneumoniae induces autophagy through the inhibition of the PI3K-I/Akt/mTOR pathway and ROS hypergeneration in A549 cells. PLoS ONE 2015, 10, e0122753. [Google Scholar] [CrossRef]
- Tattoli, I.; Philpott, D.J.; Girardin, S.E. The bacterial and cellular determinants controlling the recruitment of mTOR to the Salmonella-containing vacuole. Biol. Open 2012, 1, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Liakopoulos, A.; Mavroidi, A.; Vourli, S.; Panopoulou, M.; Zachariadou, L.; Chatzipanagiotou, S.; Spiliopoulou, I.; Zerva, L.; Petinaki, E. Molecular characterization of Streptococcus agalactiae from vaginal colonization and neonatal infections: A 4-year multicenter study in Greece. Diagn. Microbiol. Infect. Dis. 2014, 78, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Oster, C.G.; Mayer, M.M.; Avery, M.L.; Audus, K.L. Characterization of the A549 cell line as a type II pulmonary epithelial cell model for drug metabolism. Exp. Cell Res. 1998, 243, 359–366. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pantazi, I.; Papafragkos, I.; Kolliniati, O.; Lapi, I.; Tsatsanis, C.; Vergadi, E. Akt Inhibition Promotes Autophagy and Clearance of Group B Streptococcus from the Alveolar Epithelium. Pathogens 2022, 11, 1134. https://doi.org/10.3390/pathogens11101134
Pantazi I, Papafragkos I, Kolliniati O, Lapi I, Tsatsanis C, Vergadi E. Akt Inhibition Promotes Autophagy and Clearance of Group B Streptococcus from the Alveolar Epithelium. Pathogens. 2022; 11(10):1134. https://doi.org/10.3390/pathogens11101134
Chicago/Turabian StylePantazi, Ioanna, Iosif Papafragkos, Ourania Kolliniati, Ioanna Lapi, Christos Tsatsanis, and Eleni Vergadi. 2022. "Akt Inhibition Promotes Autophagy and Clearance of Group B Streptococcus from the Alveolar Epithelium" Pathogens 11, no. 10: 1134. https://doi.org/10.3390/pathogens11101134