
Genetic Pathogenesis of Inflammation-Associated Cancers in Digestive Organs


{kind=link}
{kind=link}
Abstract
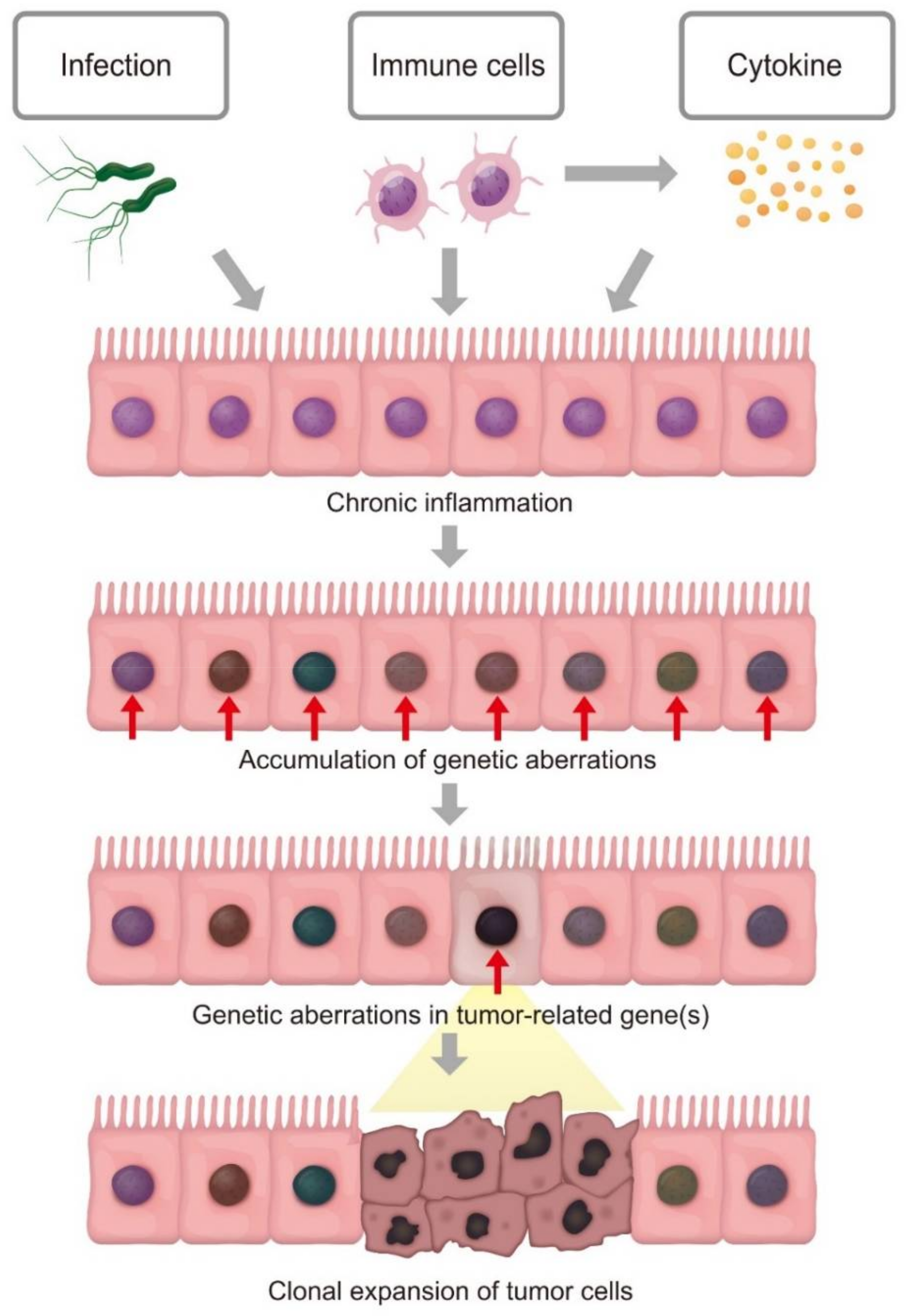
:1. Introduction
2. Colitis-Associated Colorectal Cancer
2.1. Clinical Feature and Risk Factors
2.2. Genetic Alterations Accumulated in Inflamed Epithelial Cells
3. Gastric Cancer Associated with Helicobacter pylori Infection
3.1. Clinical Feature and Risk Factors
3.2. Genetic Alterations Accumulated in Inflamed Epithelial Cells
4. Esophageal Cancer Associated with GERD
4.1. Clinical Feature and Risk Factors
4.2. Genetic Alterations Accumulated in Inflamed Epithelial Cells
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Perwez Hussain, S.; Harris, C.C. Inflammation and Cancer: An Ancient Link with Novel Potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef]
- Globocan Observatory, W. Cancer Today-World. Int. Agency Res. Cancer 2019, 876, 2018–2019. [Google Scholar]
- Beaugerie, L.; Itzkowitz, S.H. Cancers Complicating Inflammatory Bowel Disease. N. Engl. J. Med. 2015, 372, 1441–1452. [Google Scholar] [CrossRef]
- Ekbom, A.; Helmick, C.; Zack, M.; Adami, H.O. Ulcerative Colitis and Colorectal Cancer. A Population-Based Study. N. Engl. J. Med. 1990, 323, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Jess, T.; Simonsen, J.; Jorgensen, K.T.; Pedersen, B.V.; Nielsen, N.M.; Frisch, M. Decreasing Risk of Colorectal Cancer in Patients with Inflammatory Bowel Disease over 30 Years. Gastroenterology 2012, 143, 375–381.e1. [Google Scholar] [CrossRef] [PubMed]
- Eaden, J.A. The Risk of Colorectal Cancer in Ulcerative Colitis: A Meta-Analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Magro, F.; Gionchetti, P.; Eliakim, R.; Ardizzone, S.; Armuzzi, A.; Barreiro-de Acosta, M.; Burisch, J.; Gecse, K.B.; Hart, A.L.; Hindryckx, P.; et al. Third European Evidence-Based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, Diagnosis, Extra-Intestinal Manifestations, Pregnancy, Cancer Surveillance, Surgery, and Ileo-Anal Pouch Disorders. J. Crohn’s Colitis 2017, 11, 649–670. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Smits, A.M.M.; Bos, J.L. Genetic Alterations during Colorectal-Tumor Development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased P53 Mutation Load in Noncancerous Colon Tissue from Ulcerative Colitis: A Cancer-Prone Chronic Inflammatory Disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar] [PubMed]
- Yin, J.; Harpaz, N.; Tong, Y.; Huang, Y.; Laurin, J.; Greenwald, B.D.; Hontanosas, M.; Newkirk, C.; Meltzer, S.J. P53 Point Mutations in Dysplastic and Cancerous Ulcerative Colitis Lesions. Gastroenterology 1993, 104, 1633–1639. [Google Scholar] [CrossRef]
- Kern, S.E.; Redston, M.; Seymour, A.B.; Caldas, C.; Powell, S.M.; Kornacki, S.; Kinzler, K.W. Molecular Genetic Profiles of Colitis-Associated Neoplasms. Gastroenterology 1994, 107, 420–428. [Google Scholar] [CrossRef]
- Leedham, S.J.; Graham, T.A.; Oukrif, D.; McDonald, S.A.C.; Rodriguez-Justo, M.; Harrison, R.F.; Shepherd, N.A.; Novelli, M.R.; Jankowski, J.A.Z.; Wright, N.A. Clonality, Founder Mutations, and Field Cancerization in Human Ulcerative Colitis-Associated Neoplasia. Gastroenterology 2009, 136, 542–550.e6. [Google Scholar] [CrossRef] [PubMed]
- Redston, M.S.; Papadopoulos, N.; Caldas, C.; Kinzler, K.W.; Kern, S.E. Common Occurrence of APC and K-Ras Gene Mutations in the Spectrum of Colitis-Associated Neoplasias. Gastroenterology 1995, 108, 383–392. [Google Scholar] [CrossRef]
- Burmer, G.C.; Rabinovitch, P.S.; Haggitt, R.C.; Crispin, D.A.; Brentnall, T.A.; Kolli, V.R.; Stevens, A.C.; Rubin, C.E. Neoplastic Progression in Ulcerative Colitis: Histology, DNA Content, and Loss of a P53 Allele. Gastroenterology 1992, 103, 1602–1610. [Google Scholar] [CrossRef]
- Kimura, H.; Hokari, R.; Miura, S.; Shigematsu, T.; Hirokawa, M.; Akiba, Y.; Kurose, I.; Higuchi, H.; Fujimori, H.; Tsuzuki, Y.; et al. Increased Expression of an Inducible Isoform of Nitric Oxide Synthase and the Formation of Peroxynitrite in Colonic Mucosa of Patients with Active Ulcerative Colitis. Gut 1998, 42, 180–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachmilewitz, D.; Stamler, J.S.; Bachwich, D.; Karmeli, F.; Ackerman, Z.; Podolsky, D.K. Enhanced Colonic Nitric Oxide Generation and Nitric Oxide Synthase Activity in Ulcerative Colitis and Crohn’s Disease. Gut 1995, 36, 718–723. [Google Scholar] [CrossRef] [Green Version]
- Shibutani, S.; Takeshita, M.; Grollman, A.P. Insertion of Specific Bases during DNA Synthesis Past the Oxidation-Damaged Base 8-OxodG. Nature 1991, 349, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, J.L.; Wishnok, J.S.; Tannenbaum, S.R. Nitric Oxide-Induced Deamination of Cytosine and Guanine in Deoxynucleosides and Oligonucleotides. J. Biol. Chem. 1998, 273, 12689–12695. [Google Scholar] [CrossRef] [Green Version]
- Juedes, M.J.; Wogan, G.N. Peroxynitrite-Induced Mutation Spectra of PSP189 Following Replication in Bacteria and in Human Cells. Mutat. Res. Mol. Mech. Mutagen. 1996, 349, 51–61. [Google Scholar] [CrossRef]
- Cascalho, M. Advantages and Disadvantages of Cytidine Deamination. J. Immunol. 2004, 172, 6513–6518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Honjo, T.; Kinoshita, K.; Muramatsu, M. Molecular Mechanism of Class Switch Recombination: Linkage with Somatic Hypermutation. Annu. Rev. Immunol. 2002, 20, 165–196. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Marusawa, H.; Kou, T.; Nakase, H.; Fujii, S.; Fujimori, T.; Kinoshita, K.; Honjo, T.; Chiba, T. Activation-Induced Cytidine Deaminase Links Between Inflammation and the Development of Colitis-Associated Colorectal Cancers. Gastroenterology 2008, 135, 889–898. [Google Scholar] [CrossRef]
- Takai, A.; Marusawa, H.; Minaki, Y.; Watanabe, T.; Nakase, H.; Kinoshita, K.; Tsujimoto, G.; Chiba, T. Targeting Activation-Induced Cytidine Deaminase Prevents Colon Cancer Development despite Persistent Colonic Inflammation. Oncogene 2012, 31, 1733–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakiuchi, N.; Yoshida, K.; Uchino, M.; Kihara, T.; Akaki, K.; Inoue, Y.; Kawada, K.; Nagayama, S.; Yokoyama, A.; Yamamoto, S.; et al. Frequent Mutations That Converge on the NFKBIZ Pathway in Ulcerative Colitis. Nature 2020, 577, 260–265. [Google Scholar] [CrossRef]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter Pylori Infection and the Development of Gastric Cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- IARC. Schistosomes, Liver Flukes and Helicobacter Pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 1–241. [Google Scholar]
- Hatakeyama, M. Malignant Helicobacter Pylori-Associated Diseases: Gastric Cancer and MALT Lymphoma. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1149, pp. 135–149. [Google Scholar] [CrossRef]
- Webb, P.M.; Law, M.; Varghese, C.; Forman, D. Gastric Cancer and Helicobacter Pylori: A Combined Analysis of 12 Case Control Studies Nested within Prospective Cohorts. Gut 2001, 49, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Fukase, K.; Kato, M.; Kikuchi, S.; Inoue, K.; Uemura, N.; Okamoto, S.; Terao, S.; Amagai, K.; Hayashi, S.; Asaka, M. Effect of Eradication of Helicobacter Pylori on Incidence of Metachronous Gastric Carcinoma after Endoscopic Resection of Early Gastric Cancer: An Open-Label, Randomised Controlled Trial. Lancet 2008, 372, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.J.; Kook, M.C.; Kim, Y.I.; Cho, S.J.; Lee, J.Y.; Kim, C.G.; Park, B.; Nam, B.H. Helicobacter Pylori Therapy for the Prevention of Metachronous Gastric Cancer. N. Engl. J. Med. 2018, 378, 1085–1095. [Google Scholar] [CrossRef]
- Aoi, T. Risk of Subsequent Development of Gastric Cancer in Patients with Previous Gastric Epithelial Neoplasia. Gut 2006, 55, 588–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurén, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Correa, P. A Human Model of Gastric Carcinogenesis. Cancer Res. 1988, 48, 3554–3560. [Google Scholar]
- De Vries, A.C.; van Grieken, N.C.T.; Looman, C.W.N.; Casparie, M.K.; de Vries, E.; Meijer, G.A.; Kuipers, E.J. Gastric Cancer Risk in Patients With Premalignant Gastric Lesions: A Nationwide Cohort Study in The Netherlands. Gastroenterology 2008, 134, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.T.; Lee, H.; Sousa, J.F.; Weis, V.G.; O’Neal, R.L.; Finke, P.E.; Romero–Gallo, J.; Shi, G.; Mills, J.C.; Peek, R.M.; et al. Mature Chief Cells Are Cryptic Progenitors for Metaplasia in the Stomach. Gastroenterology 2010, 139, 2028–2037.e9. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Choi, E.; Petersen, C.P.; Noto, J.M.; Romero-Gallo, J.; Piazuelo, M.B.; Washington, M.K.; Peek, R.M.; Goldenring, J.R. Characterization of Progressive Metaplasia in the Gastric Corpus Mucosa of Mongolian Gerbils Infected with Helicobacter Pylori. J. Pathol. 2016, 239, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Mizuguchi, A.; Takai, A.; Shimizu, T.; Matsumoto, T.; Kumagai, K.; Miyamoto, S.; Seno, H.; Marusawa, H. Genetic Features of Multicentric/Multifocal Intramucosal Gastric Carcinoma. Int. J. Cancer 2018, 143, 1923–1934. [Google Scholar] [CrossRef] [Green Version]
- Rokutan, H.; Abe, H.; Nakamura, H.; Ushiku, T.; Arakawa, E.; Hosoda, F.; Yachida, S.; Tsuji, Y.; Fujishiro, M.; Koike, K.; et al. Initial and Crucial Genetic Events in Intestinal-Type Gastric Intramucosal Neoplasia. J. Pathol. 2019, 247, 494–504. [Google Scholar] [CrossRef]
- Shimizu, T.; Marusawa, H.; Matsumoto, Y.; Inuzuka, T.; Ikeda, A.; Fujii, Y.; Minamiguchi, S.; Miyamoto, S.; Kou, T.; Sakai, Y.; et al. Accumulation of Somatic Mutations in TP53 in Gastric Epithelium with Helicobacter Pylori Infection. Gastroenterology 2014, 147, 407–417.e3. [Google Scholar] [CrossRef]
- Huang, K.K.; Ramnarayanan, K.; Zhu, F.; Srivastava, S.; Xu, C.; Tan, A.L.K.; Lee, M.; Tay, S.; Das, K.; Xing, M.; et al. Genomic and Epigenomic Profiling of High-Risk Intestinal Metaplasia Reveals Molecular Determinants of Progression to Gastric Cancer. Cancer Cell 2018, 33, 137–150.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Marusawa, H.; Kinoshita, K.; Endo, Y.; Kou, T.; Morisawa, T.; Azuma, T.; Okazaki, I.M.; Honjo, T.; Chiba, T. Helicobacter Pylori Infection Triggers Aberrant Expression of Activation-Induced Cytidine Deaminase in Gastric Epithelium. Nat. Med. 2007, 13, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, N.; Akiyama, J.; Marusawa, H.; Shimbo, T.; Liu, Y.; Igari, T.; Nakashima, R.; Watanabe, H.; Uemura, N.; Chiba, T. Enhanced Expression of Activation-Induced Cytidine Deaminase in Human Gastric Mucosa Infected by Helicobacter Pylori and Its Decrease Following Eradication. J. Gastroenterol. 2014, 49, 427–435. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Marusawa, H.; Kinoshita, K.; Niwa, Y.; Sakai, Y.; Chiba, T. Up-Regulation of Activation-Induced Cytidine Deaminase Causes Genetic Aberrations at the CDKN2b-CDKN2a in Gastric Cancer. Gastroenterology 2010, 139, 1984–1994. [Google Scholar] [CrossRef] [Green Version]
- Gholipour, C.; Shalchi, R.A.; Abbasi, M. A Histopathological Study of Esophageal Cancer on the Western Side of the Caspian Littoral from 1994 to 2003. Dis. Esophagus 2008, 21, 322–327. [Google Scholar] [CrossRef]
- Tran, G.D.; Di Sun, X.; Abnet, C.C.; Fan, J.H.; Dawsey, S.M.; Dong, Z.W.; Mark, S.D.; Qiao, Y.L.; Taylor, P.R. Prospective Study of Risk Factors for Esophageal and Gastric Cancers in the Linxian General Population Trial Cohort in China. Int. J. Cancer 2005, 113, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Engel, L.S.; Chow, W.H.; Vaughan, T.L.; Gammon, M.D.; Risch, H.A.; Stanford, J.L.; Schoenberg, J.B.; Mayne, S.T.; Dubrow, R.; Rotterdam, H.; et al. Population Attributable Risks of Esophageal and Gastric Cancers. J. Natl. Cancer Inst. 2003, 95, 1404–1413. [Google Scholar] [CrossRef]
- Dikken, J.L.; Lemmens, V.E.; Wouters, M.W.J.M.; Wijnhoven, B.P.; Siersema, P.D.; Nieuwenhuijzen, G.A.; Van Sandick, J.W.; Cats, A.; Verheij, M.; Coebergh, J.W.; et al. Increased Incidence and Survival for Oesophageal Cancer but Not for Gastric Cardia Cancer in the Netherlands. Eur. J. Cancer 2012, 48, 1624–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcedo, J.; Ferrández, A.; Arenas, J.; Sopeña, F.; Ortego, J.; Sainz, R.; Lanas, A. Trends in Barrett’s Esophagus Diagnosis in Southern Europe: Implications for Surveillance. Dis. Esophagus 2009, 22, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.B.; Reed, C.E. Epidemiology of Esophageal Cancer. Surg. Clin. N. Am. 2012, 92, 1077–1087. [Google Scholar] [CrossRef]
- Morita, S.; Matsumoto, Y.; Okuyama, S.; Ono, K.; Kitamura, Y.; Tomori, A.; Oyama, T.; Amano, Y.; Kinoshita, Y.; Chiba, T.; et al. Bile Acid-Induced Expression of Activation-Induced Cytidine Deaminase during the Development of Barrett’s Oesophageal Adenocarcinoma. Carcinogenesis 2011, 32, 1706–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T. The Nuclear Factor NF-B Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Tak, P.P.; Firestein, G.S. NF-ΚB: A Key Role in Inflammatory Diseases. J. Clin. Invest. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Miwa, K.; Sahara, H.; Segawa, M.; Kinami, S.; Sato, T.; Miyazaki, I.; Hattori, T. Reflux of Duodenal or Gastro-Duodenal Contents Induces Esophageal Carcinoma in Rats. Int. J. Cancer 1996, 67, 269–274. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Lada, A.G.; Goncearenco, A.; Green, M.R.; De, S.; Nudelman, G.; Panchenko, A.R.; Koonin, E.V.; Pavlov, Y.I. Activation Induced Deaminase Mutational Signature Overlaps with CpG Methylation Sites in Follicular Lymphoma and Other Cancers. Sci. Rep. 2016, 6, 38133. [Google Scholar] [CrossRef] [Green Version]
- Wink, D.; Kasprzak, K.; Maragos, C.; Elespuru, R.; Misra, M.; Dunams, T.; Cebula, T.; Koch, W.; Andrews, A.; Allen, J.; et al. DNA Deaminating Ability and Genotoxicity of Nitric Oxide and Its Progenitors. Science 1991, 254, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Clemons, N.J.; McColl, K.E.L.; Fitzgerald, R.C. Nitric Oxide and Acid Induce Double-Strand DNA Breaks in Barrett’s Esophagus Carcinogenesis via Distinct Mechanisms. Gastroenterology 2007, 133, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired Exome Analysis of Barrett’s Esophagus and Adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, J.M.J.; Ross-Innes, C.S.; Shannon, N.; Lynch, A.G.; Forshew, T.; Barbera, M.; Murtaza, M.; Ong, C.-A.J.; Lao-Sirieix, P.; Dunning, M.J.; et al. Ordering of Mutations in Preinvasive Disease Stages of Esophageal Carcinogenesis. Nat. Genet. 2014, 46, 837–843. [Google Scholar] [CrossRef]
- Galipeau, P.C.; Oman, K.M.; Paulson, T.G.; Sanchez, C.A.; Zhang, Q.; Marty, J.A.; Delrow, J.J.; Kuhner, M.K.; Vaughan, T.L.; Reid, B.J.; et al. NSAID Use and Somatic Exomic Mutations in Barrett’s Esophagus. Genome Med. 2018, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and Whole-Genome Sequencing of Esophageal Adenocarcinoma Identifies Recurrent Driver Events and Mutational Complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Killcoyne, S.; Gregson, E.; Wedge, D.C.; Woodcock, D.J.; Eldridge, M.D.; de la Rue, R.; Miremadi, A.; Abbas, S.; Blasko, A.; Kosmidou, C.; et al. Genomic Copy Number Predicts Esophageal Cancer Years before Transformation. Nat. Med. 2020, 26, 1726–1732. [Google Scholar] [CrossRef]
- Morgan, R.L.; Baack, B.; Smith, B.D.; Yartel, A.; Pitasi, M.; Falck-Ytter, Y. Eradication of Hepatitis C Virus Infection and the Development of Hepatocellular Carcinoma: A Meta-Analysis of Observational Studies. Ann. Intern. Med. 2013, 158, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahon, P.; Bourcier, V.; Layese, R.; Audureau, E.; Cagnot, C.; Marcellin, P.; Guyader, D.; Fontaine, H.; Larrey, D.; De Lédinghen, V.; et al. Eradication of Hepatitis C Virus Infection in Patients With Cirrhosis Reduces Risk of Liver and Non-Liver Complications. Gastroenterology 2017, 152, 142–156.e2. [Google Scholar] [CrossRef] [Green Version]
- Morgan, T.R.; Ghany, M.G.; Kim, H.Y.; Snow, K.K.; Shiffman, M.L.; De Santo, J.L.; Lee, W.M.; Di Bisceglie, A.M.; Bonkovsky, H.L.; Dienstag, J.L.; et al. Outcome of Sustained Virological Responders with Histologically Advanced Chronic Hepatitis C. Hepatology 2010, 52, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakanishi, R.; Shimizu, T.; Kumagai, K.; Takai, A.; Marusawa, H. Genetic Pathogenesis of Inflammation-Associated Cancers in Digestive Organs. Pathogens 2021, 10, 453. https://doi.org/10.3390/pathogens10040453
Nakanishi R, Shimizu T, Kumagai K, Takai A, Marusawa H. Genetic Pathogenesis of Inflammation-Associated Cancers in Digestive Organs. Pathogens. 2021; 10(4):453. https://doi.org/10.3390/pathogens10040453
Chicago/Turabian StyleNakanishi, Risa, Takahiro Shimizu, Ken Kumagai, Atsushi Takai, and Hiroyuki Marusawa. 2021. "Genetic Pathogenesis of Inflammation-Associated Cancers in Digestive Organs" Pathogens 10, no. 4: 453. https://doi.org/10.3390/pathogens10040453