
Adjustment of Matrix Effects in Analysis of 36 Secondary Metabolites of Microbial and Plant Origin in Indoor Floor Dust Using Liquid Chromatography-Tandem Mass Spectrometry


Abstract
:1. Introduction
2. Materials and Methods
2.1. Standard Materials and Chemicals
2.2. Preparation of Standard Solutions for Spiking to Dust
2.3. Preparation of External Calibration Curves
2.4. Preparation of Test Dust and Extraction of Metabolites from the Spiked Dust
2.5. Chromatographic Conditions
2.6. MS Parameters and Transitions
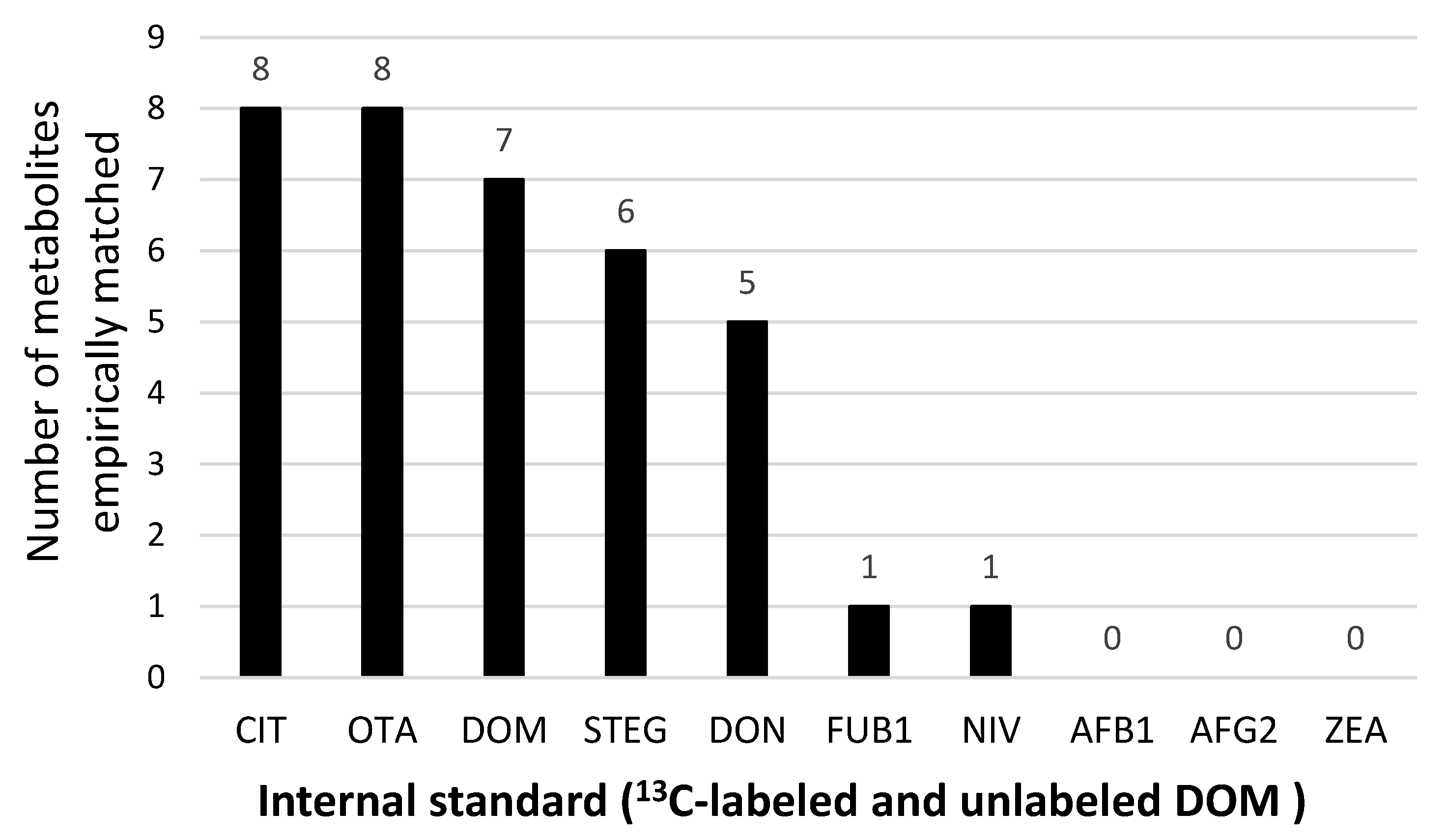
2.7. Selection of Best-Performing ISTD for Each Metabolite and Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Dillon, H.K.; Miller, J.D.; Sorenson, W.G.; Douwes, J.; Jacobs, R.R. Review of methods applicable to the assessment of mold exposure to children. Environ. Health Perspect. 1999, 107, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Andersen, B.; Frisvad, J.C.; Søndergaard, I.; Rasmussen, I.S.; Larsen, L.S. Associations between fungal species and water-damaged building materials. Appl. Environ. Microbiol. 2011, 77, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Who Guidelines for Indoor Air Quality: Dampness and Mould; Druckpartner Moser: Rheinbach, Germany, 2009; Available online: https://www.euro.who.int/__data/assets/pdf_file/0017/43325/E92645.pdf (accessed on 10 February 2023).
- Mendell, M.J.; Mirer, A.G.; Cheung, K.; Tong, M.; Douwes, J. Respiratory and allergic health effects of dampness, mold, and dampness-related agents: A review of the epidemiologic evidence. Environ. Health Perspect. 2011, 119, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Cox-Ganser, J.M. Mold exposure and respiratory health in damp indoor environments. Front. Biosci. 2011, E3, 757–771. [Google Scholar] [CrossRef]
- Saito, R.; Park, J.-H.; LeBouf, R.; Green, B.J.; Park, Y. Measurement of macrocyclic trichothecene in floor dust of water-damaged buildings using gas chromatography/tandem mass spectrometry—Dust matrix effects. J. Occup. Environ. Hyg. 2016, 13, 442–450. [Google Scholar] [CrossRef]
- Cai, G.-H.; Hashim, J.H.; Hashim, Z.; Ali, F.; Bloom, E.; Larsson, L.; Lampa, E.; Norbäck, D. Fungal DNA, allergens, mycotoxins and associations with asthmatic symptoms among pupils in schools from johor bahru, malaysia. Pediatr. Allergy Immunol. 2011, 22, 290–297. [Google Scholar] [CrossRef]
- Kirjavainen, P.V.; Täubel, M.; Karvonen, A.M.; Sulyok, M.; Tiittanen, P.; Krska, R.; Hyvärinen, A.; Pekkanen, J. Microbial secondary metabolites in homes in association with moisture damage and asthma. Indoor Air 2015, 26, 448–456. [Google Scholar] [CrossRef]
- Dahal, U.P.; Jones, J.P.; Davis, J.A.; Rock, D.A. Small molecule quantification by liquid chromatography-mass spectrometry for metabolites of drugs and drug candidates. Drug Metab. Dispos. 2011, 39, 2355–2360. [Google Scholar] [CrossRef]
- Jaderson, M.; Park, J.-H. Evaluation of matrix effects in quantifying microbial secondary metabolites in indoor dust using ultraperformance liquid chromatograph–tandem mass spectrometer. Saf. Health Work. 2019, 10, 196–204. [Google Scholar] [CrossRef]
- Benijts, T.; Dams, R.; Lambert, W.; De Leenheer, A. Countering matrix effects in environmental liquid chromatography–electrospray ionization tandem mass spectrometry water analysis for endocrine disrupting chemicals. J. Chromatogr. A 2004, 1029, 153–159. [Google Scholar] [CrossRef]
- Rimayi, C.; Odusanya, D.; Mtunzi, F.; Tsoka, S. Alternative calibration techniques for counteracting the matrix effects in GC–MS-SPE pesticide residue analysis—A statistical approach. Chemosphere 2015, 118, 35–43. [Google Scholar] [CrossRef]
- Fabregat-Cabello, N.; Zomer, P.; Sancho, J.V.; Roig-Navarro, A.F.; Mol, H.G.J. Comparison of approaches to deal with matrix effects in lc-ms/ms based determinations of mycotoxins in food and feed. World Mycotoxin J. 2016, 9, 149–161. [Google Scholar] [CrossRef]
- Steiner, D.; Krska, R.; Malachová, A.; Taschl, I.; Sulyok, M. Evaluation of matrix effects and extraction efficiencies of lc–ms/ms methods as the essential part for proper validation of multiclass contaminants in complex feed. J. Agric. Food Chem. 2020, 68, 3868–3880. [Google Scholar] [CrossRef]
- Vishwanath, V.; Sulyok, M.; Labuda, R.; Bicker, W.; Krska, R. Simultaneous determination of 186 fungal and bacterial metabolites in indoor matrices by liquid chromatography/tandem mass spectrometry. Anal. Bioanal. Chem. 2009, 395, 1355–1372. [Google Scholar] [CrossRef]
- Park, J.-H.; Cox-Ganser, J.; Rao, C.; Kreiss, K. Fungal and endotoxin measurements in dust associated with respiratory symptoms in a water-damaged office building. Indoor Air 2006, 16, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Cox-Ganser, J.M.; White, S.K.; Laney, A.S.; Caulfield, S.M.; Turner, W.A.; Sumner, A.D.; Kreiss, K. Bacteria in a water-damaged building: Associations of actinomycetes and non-tuberculous mycobacteria with respiratory health in occupants. Indoor Air 2016, 27, 24–33. [Google Scholar] [CrossRef]
- Delmulle, B.; De Saeger, S.; Adams, A.; De Kimpe, N.; Van Peteghem, C. Development of a liquid chromatography/tandem mass spectrometry method for the simultaneous determination of 16 mycotoxins on cellulose filters and in fungal cultures. Rapid Commun. Mass Spectrom. 2006, 20, 771–776. [Google Scholar] [CrossRef]
- Ediage, E.N.; Di Mavungu, J.D.; Monbaliu, S.; Van Peteghem, C.; De Saeger, S. A validated multianalyte lc–ms/ms method for quantification of 25 mycotoxins in cassava flour, peanut cake and maize samples. J. Agric. Food Chem. 2011, 59, 5173–5180. [Google Scholar] [CrossRef]
- Choi, B.K.; Hercules, D.M.; Gusev, A.I. Lc-ms/ms signal suppression effects in the analysis of pesticides in complex environmental matrices. Fresenius J. Anal. Chem. 2001, 369, 370–377. [Google Scholar] [CrossRef]
- Marginean, I.; Kelly, R.T.; Moore, R.J.; Prior, D.C.; LaMarche, B.L.; Tang, K.; Smith, R.D. Selection of the optimum electrospray voltage for gradient elution lc-ms measurements. J. Am. Soc. Mass Spectrom. 2008, 20, 682–688. [Google Scholar] [CrossRef]
- Banerjee, S.; Mazumdar, S. Electrospray ionization mass spectrometry: A technique to access the information beyond the molecular weight of the analyte. Int. J. Anal. Chem. 2012, 2012, 282574. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.A.; Chan, H.P. 8—Adhesive technology for photonics. In Advanced Adhesives in Electronics; Alam, M.O., Bailey, C., Eds.; Woodhead Publishing: Cambridge, UK, 2011; pp. 214–258. [Google Scholar] [CrossRef]
- Fenn, J.B. Ion formation from charged droplets: Roles of geometry, energy, and time. J. Am. Soc. Mass Spectrom. 1993, 4, 524–535. [Google Scholar] [CrossRef]
- Nguyen, S.; Fenn, J.B. Gas-phase ions of solute species from charged droplets of solutions. Proc. Natl. Acad. Sci. USA 2007, 104, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Page, J.S.; Kelly, R.T.; Tang, K.; Smith, R.D. Ionization and transmission efficiency in an electrospray ionization–mass spectrometry interface. J. Am. Soc. Mass Spectrom. 2007, 18, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Skillman, B.; Kerrigan, S. Identification of suvorexant in blood using lc–ms-ms: Important considerations for matrix effects and quantitative interferences in targeted assays. J. Anal. Toxicol. 2019, 44, 245–255. [Google Scholar] [CrossRef]
- Tisler, S.; Pattison, D.I.; Christensen, J. H Correction of matrix effects for reliable non-target screening lc–esi–ms analysis of wastewater. Anal. Chem. 2021, 93, 8432–8441. [Google Scholar] [CrossRef]
- Spanjer, M.C.; Rensen, P.M.; Scholten, J.M. Lc–ms/ms multi-method for mycotoxins after single extraction, with validation data for peanut, pistachio, wheat, maize, cornflakes, raisins and figs. Food Addit. Contam. Part A 2008, 25, 472–489. [Google Scholar] [CrossRef]
- Abbas, H.K.; Cartwright, R.D.; Xie, W.; Mirocha, C.J.; Richard, J.L.; Dvorak, T.J.; Sciumbato, G.L.; Shier, W.T. Mycotoxin production by fusarium proliferatum isolates from rice with fusarium sheath rot disease. Mycopathologia 1999, 147, 97–104. [Google Scholar] [CrossRef]
- Hübner, F.; Harrer, H.; Fraske, A.; Kneifel, S.; Humpf, H.-U. Large scale purification of b-type fumonisins using centrifugal partition chromatography (cpc). Mycotoxin Res. 2011, 28, 37–43. [Google Scholar] [CrossRef]
- Sulyok, M.; Krska, R.; Schuhmacher, R. A liquid chromatography/tandem mass spectrometric multi-mycotoxin method for the quantification of 87 analytes and its application to semi-quantitative screening of moldy food samples. Anal. Bioanal. Chem. 2007, 389, 1505–1523. [Google Scholar] [CrossRef]
- European Union. Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed Sante 11312/2021; European Union: Maastricht, The Netherlands, 2022; Volume 20. [Google Scholar]
- Stahnke, H.; Reemtsma, T.; Alder, L. Compensation of matrix effects by postcolumn infusion of a monitor substance in multiresidue analysis with LC−MS/MS. Anal. Chem. 2009, 81, 2185–2192. [Google Scholar] [CrossRef]
- Akkreditierungsstelle, D. Anforderungen an Laboratorien im Gesundheitlichen Verbraucherschutz- Wirkstoff–Multimethoden zur Pestizidanalytik in Lebens-und Futtermitteln; Revision: 1.4; Deutsche Akkreditierungsstelle: Berlin, Germany, 2017; Volume 11, Available online: http://docplayer.org/44255530-Anforderungen-an-laboratorien-im-gesundheitlichen-verbraucherschutz-wirkstoff-multimethoden-zur-pestizidanalytik-in-lebens-und-futtermitteln.html (accessed on 10 February 2023).
- Nasiri, A.; Jahani, R.; Mokhtari, S.; Yazdanpanah, H.; Daraei, B.; Faizi, M.; Kobarfard, F. Overview, consequences, and strategies for overcoming matrix effects in lc-ms analysis: A critical review. Analyst 2021, 146, 6049–6063. [Google Scholar] [CrossRef]


{kind=link}
| No. | Analyte (SM) | Abbreviation | Supplier * | CAS no. |
|---|---|---|---|---|
| 1 | 3-Nitropropionic acid | NITP | Sigma-Aldrich | 504-88-1 |
| 2 | Aflatoxin B1 | AFB1 | Sigma-Aldrich | 1162-65-8 |
| 3 | Aflatoxin B2 | AFB2 | Fermentek | 7220-81-7 |
| 4 | Aflatoxin G1 | AFG1 | Sigma-Aldrich | 1165-39-5 |
| 5 | Aflatoxin G2 | AFG2 | Sigma-Aldrich | 7241-98-7 |
| 6 | Alternariol | ALT | Sigma-Aldrich | 641-38-3 |
| 7 | Alternariol monomethylether | AME | Adipogen | 26894-49-5 |
| 8 | Asperglaucide | ASPG | ChemFaces | 56121-42-7 |
| 9 | Asperphenamate | ASPH | Cayman Chemical | 63631-36-7 |
| 10 | Chaetoglobosin A | CTGA | Adipogen | 50335-03-0 |
| 11 | Citreorosein | CITRO | ChemFaces | 481-73-2 |
| 12 | Citrinin | CIT | Sigma-Aldrich | 518-75-2 |
| 13 | Cyclo (L-Pro-L-Tyr) | CYCT | Bioaustralis | 4549-02-4 |
| 14 | Cyclo (L-Pro-L-Val) | CYCV | Cayman Chemical | 2854-40-2 |
| 15 | Deoxynivalenol | DON | Sigma-Aldrich | 51481-10-8 |
| 16 | Emodin | EMOD | Sigma-Aldrich | 518-82-1 |
| 17 | Enniatin B | ENNB | Cayman Chemical | 917-13-5 |
| 18 | Enniatin B1 | ENNB1 | Cayman Chemical | 19914-20-6 |
| 19 | Fumonisin B1 | FUB1 | Sigma-Aldrich | 116355-83-0 |
| 20 | Integracin A | INTA | Santa Cruz | 224186-03-2 |
| 21 | Integracin B | INTB | Santa Cruz | 224186-05-4 |
| 22 | Linamarin | LIN | Cayman Chemical | 554-35-8 |
| 23 | Lotaustralin | LOT | Sigma-Aldrich | 534-67-8 |
| 24 | N-Benzoyl-L-phenylalanine | NBLP | Sigma-Aldrich | 2566-22-5 |
| 25 | Neoechinulin A | NEOA | ChemFaces | 51551-29-2 |
| 26 | Nivalenol | NIV | Fermentek | 23282-20-4 |
| 27 | Ochratoxin A | OTA | Sigma-Aldrich | 303-47-9 |
| 28 | Roquefortine C | ROQC | Santa Cruz | 58735-64-1 |
| 29 | Skyrin | SKY | Sigma-Aldrich | 602-06-2 |
| 30 | Stachybotrylactam | STCH | Santa Cruz | 163391-76-2 |
| 31 | Sterigmatocystin | STEG | Sigma-Aldrich | 10048-13-2 |
| 32 | Usnic Acid | USN | Cayman Chemical | 125-46-2 |
| 33 | Valinomycin | VAL | Sigma-Aldrich | 2001-95-8 |
| 34 | Verrucarin A | VERA | Sigma-Aldrich | 3148-09-2 |
| 35 | Verrucarol | VERO | Sigma-Aldrich | 2198-92-7 |
| 36 | Zearalenone | ZEA | Sigma-Aldrich | 17924-92-4 |
| No. | Analyte | Abbreviation | Supplier * | CAS no. |
|---|---|---|---|---|
| 1 | 13C-aflatoxin B1 | 13C-AFB1 | Romer Labs | 1217449-45-0 |
| 2 | 13C-aflatoxin G2 | 13C-AFG2 | Romer Labs | 1217462-49-1 |
| 3 | 13C-citrinin | 13C-CIT | Romer Labs | ** 518-75-2 (unlabeled) |
| 4 | 13C-deoxynivalenol | 13C-DON | Romer Labs | 911392-36-4 |
| 5 | 13C-fumonisin B1 | 13C-FUB1 | Romer Labs | 1217458-62-2 |
| 6 | 13C-nivalenol | 13C-NIV | Romer Labs | 911392-40-0 |
| 7 | 13C-ochratoxin A | 13C-OTA | Romer Labs | 911392-42-2 |
| 8 | 13C-sterigmatocystin | 13C-STEG | Romer Labs | ** 10048-13-2 (unlabeled) |
| 9 | 13C-zearalenone | 13C-ZEA | Romer Labs | 911392-43-3 |
| 10 | Deepoxy-deoxynivalenol | DOM | Sigma-Aldrich | 88054-24-4 |
| Metabolite/ISTD | RT (min) | Precursor Ion (m/z) | m/z of Product Ion #1 | m/z of Product Ion #2 | Cone Voltage (V) |
|---|---|---|---|---|---|
| NITP (neg) | 2.05 | 117.8 [M-H] − | 45.89 (6) | –* | 20 |
| LIN | 2.8 | 265.2 [M + NH4] + | 163.08 (10) | 85.03 (20) | 34 |
| NIV | 3.68 | 313.22 [M + H] + | 125.01 (12) | 205.06 (12) | 25 |
| 13C-NIV | 3.68 | 328.20 [M + H] + | 217.08 (12) | 186.04 (12) | 26 |
| CYCT | 4.07 | 261.09 [M + H] + | 135.98 (18) | 28 (106.99) | 34 |
| DON | 4.14 | 297.10 [M + H] + | 249.10 (10) | 203.07 (14) | 28 |
| 13C-DON (pos) | 4.14 | 312.10 [M + H] + | 216.20 (16) | 263.20 (17) | 26 |
| 13C-DON (neg) ** | 4.14 | 310.05 [M-H] − | 261.07 (10) | 279.08 (10) | 38 |
| CYCV | 4.31 | 197.03 [M + H] + | 69.98 (22) | 169.10 (14) | 38 |
| DOM (pos) | 4.51 | 281.16 [M + H] + | 109.00 (22) | 233.11 (10) | 26 |
| DOM (neg) ** | 4.51 | 339.23 [M-H] − | 249.05 (10) | 279.12 (12) | 20 |
| VERO | 4.7 | 267.12 [M + H] + | 249.15 (8) | 231.07 (10) | 14 |
| AFG2 | 4.83 | 331.04 [M + H] + | 313.05 (26) | 245.05 (30) | 50 |
| 13C-AFG2 | 4.83 | 348.10 [M + H] + | 259.00 (32) | 330.00 (36) | 56 |
| AFG1 | 4.96 | 329.03 [M + H] + | 243.05 (28) | 199.88 (40) | 50 |
| AFB2 | 5.11 | 315.05 [M + H] + | 287.05 (28) | 259.04 (28) | 54 |
| AFB1 | 5.23 | 313.10 [M + H] + | 284.86 (24) | 241.11 (40) | 62 |
| 13C-AFB1 | 5.23 | 330.10 [M + H] + | 301.00 (18) | 255.10 (26) | 54 |
| CIT | 5.44 | 251.05 [M + H] + | 233.1 (16) | 191.00 (26) | 28 |
| 13C-CIT | 5.44 | 264.01 [M + H] + | 246.05 (15) | –* | 34 |
| NBLP | 5.49 | 270.02 [M + H] + | 104.97 (18) | 119.99 (12) | 32 |
| NEOA | 5.69 | 324.06 [M + H] + | 256.05 (10) | 268.07 (12) | 24 |
| ALT | 5.74 | 258.89 [M + H] + | 185.00 (30) | 127.85 (46) | 56 |
| ASPG | 5.78 | 445.17 [M + H] + | 349.16 (18) | 107.03 (38) | 40 |
| FUB1 | 5.78 | 722.36 [M + H] + | 352.31 (36) | 74.02 (58) | 56 |
| 13C-FUB1 | 5.78 | 756.40 [M + H] + | 374.33 (38) | 356.32 (44) | 66 |
| CITRO (neg) | 6.09 | 284.95 [M-H] − | 211.01 (40) | 224.07 (32) | 66 |
| VERA | 6.11 | 520.38 [M + NH4] + | 249.09 (18) | 457.19 (14) | 24 |
| ROQC | 6.17 | 390.15 [M + H] + | 193.00 (26) | 322.12 (20) | 48 |
| OTA | 6.24 | 404.05 [M + H] + | 238.96 (22) | (358.06) 14 | 32 |
| 13C-OTA (pos) | 6.24 | 424.07 [M + H] + | 250.03 (26) | 109.94 (76) | 34 |
| 13C-OTA (neg) ** | 6.24 | 422 [M-H] − | 174.99 (40) | 377.05 (20) | 50 |
| ZEA | 6.44 | 321.16 [M + H] + | 303.13 (14) | 189.10 (20) | 20 |
| 13C-ZEA | 6.44 | 337.10 [M + H] + | 243.15 (22) | 185.07 (42) | 22 |
| CTGA | 6.59 | 529.16 [M + H] + | 130.01 (38) | 292.05 (24) | 26 |
| STEG | 6.72 | 325.03 [M + H] + | 309.99 (22) | 281.05 (34) | 56 |
| 13C-STEG | 6.72 | 343.01 [M + H] + | 327.06 (28) | 297.10 (40) | 54 |
| AME (neg) | 6.76 | 270.97 [M-H] − | 255.99 (22) | 182.98 (4) | 52 |
| ASPH | 6.97 | 507.19 [M + H] + | 238.08 (18) | 256.09 (12) | 34 |
| STCH | 7.04 | 386.19 [M + H] + | 178.05 (38) | 150.16 (50) | 66 |
| EMOD (neg) | 7.46 | 268.95 [M-H] − | 224.99 (24) | 240.92 (30) | 56 |
| ENNB | 7.55 | 640.44 [M + H] + | 196.10 (26) | 86.05 (70) | 60 |
| SKY | 7.66 | 538.99 [M + H] + | 521.08 (24) | 503.87 (40) | 64 |
| ENNB1 | 7.67 | 654.40 [M + H] + | 86.05 (62) | 196.10 (28) | 50 |
| USN (neg) | 7.67 | 343.12 [M-H] − | 328.00 (22) | 259.01 (18) | 44 |
| INTB | 7.79 | 587.36 [M + H] + | 307.15 (18) | 166.99 (26) | 24 |
| LOT | 7.9 | 262.02 [M + H] + | 84.94 (22) | 162.98 (8) | 16 |
| INTA | 8.18 | 629.37 [M + H] + | 349.18 (14) | 289.18 (28) | 28 |
| VAL | 9.13 | 1128.65 [M + NH4] + | 172.15 (78) | 343.30 (64) | 98 |
| Compound | Empirically Determined ISTD |
|---|---|
| 3-NITP | 13C-OTA |
| AFB1 * | 13C-OTA |
| AFB2 | 13C-OTA |
| AFG1 ** | 13C-OTA |
| AFG2 * | 13C-OTA |
| ALT | 13C-STEG |
| AME | 13C-DON |
| ASPG | DOM |
| ASPH | DOM |
| CTGA | 13C-OTA |
| CITRO | 13C-DON |
| CIT | 13C-CIT |
| CYCT | DOM |
| CYCV ** | 13C-STEG |
| DON *,** | 13C-OTA |
| EMOD | DOM |
| ENNB | DOM |
| ENNB1 | DOM |
| FUB1 | 13C-FUB1 |
| INTA ** | 13C-CIT |
| INTAB | 13C-DON |
| LIN | 13C-CIT |
| LOT | 13C-STEG |
| NBLP | 13C-CIT |
| NEOA | 13C-CIT |
| NIV* | 13C-CIT |
| OTA | 13C-OTA |
| ROQC | 13C-DON |
| SKY | 13C-STEG |
| STCH | 13C-CIT |
| STEG | 13C-STEG |
| USN | 13C-DON |
| VAL | 13C-NIV |
| VERA | DOM |
| VERO ** | 13C-CIT |
| ZEA * | 13C-STEG |
| Initial Experiment | First Validation Experiment | Second Validation Experiment | ||||
|---|---|---|---|---|---|---|
| Dust Sample A | Dust Sample A | Dust Sample B | ||||
| Metabolite Group | LOQ (pg/µL) | Mean (CV) | Mean (CV) | Difference in Recovery (Validation 1—Initial) | Mean (CV) | Difference in Recovery (Validation 2—Initial) |
| 1. Seventeen metabolites with acceptable average recoveries from the initial experiment | ||||||
| AFB1 | 0.8 | 107.2 (9.5) * | 97.4 (27.8) ** | −9.8 | 116.9 (27.9) ** | 9.7 |
| AFB2 | 1.3 | 96.3 (9.9) * | 107.5 (28.9) ** | 11.2 | 108.5 (24.6) ** | 12.2 |
| AFG1 | 3.9 | 87.7 (6.5) * | 54.9 (8.2) † | −32.8 | 132.3 (59.9) ** | 44.6 |
| AFG2 | 1.1 | 112.6 (9.8) * | 114.5 (29.3) ** | 1.9 | 60 (56.3) ** | −52.6 |
| ALT | 10 | 133.6 (18.1) * | -†† | - | 63.4 (60.7) ** | −70.2 |
| ASPG | 0.05 | 77.5 (20.5) * | 88.2 (15.5) * | 10.7 | 91.6 (32) ** | 14.1 |
| CIT | 3.9 | 116.3 (5.5) * | 122 (6.5) * | 5.7 | 106.1 (7.1) * | −10.2 |
| CTGA | 2.5 | 89 (8) * | 71.4 (22.8) ** | −17.6 | 53.9 (40.1) † | −35.1 |
| CYCT | 2 | 99 (18.6) * | 124.7 (19.8) * | 25.7 | 79.2 (22) ** | −19.8 |
| FUB1 | 62.5 | 101.8 (12.5) * | 13.7 (35.4) † | −88.1 | 37.5 (41.7) † | −64.3 |
| LIN | 7.5 | 67.5 (17.4) * | 50.7 (28.2) † | −16.8 | -†† | - |
| NBLP | 0.33 | 116.9 (8) * | 108.7 (12.8) * | −8.2 | 134.6 (35.8) ** | 17.7 |
| NEOA | 2.6 | 95.6 (6.3) * | 154.4 (8.7) † | 58.8 | 161.6 (172.5) † | 66 |
| NITP | 42 | 94.0 (3.9) * | -†† | - | -†† | - |
| OTA | 0.1 | 111 (9.8) * | 112.3 (25.4) ** | 1.3 | 164.1 (34.8) † | 53.1 |
| STCH | 1.3 | 88.9 (6.7) * | 90 (5.4) * | 1.1 | 53.1 (49.4) † | −35.8 |
| VERO | 31 | 69.1 (17.9) * | 40.9 (12.8) † | −28.2 | 108.8 (45.3) ** | 39.7 |
| 2. Nine metabolites with marginally acceptable average recoveries from the initial experiment | ||||||
| CYCV | 5.7 | 120.4 (59.1) ** | 189.5 (37.3) † | 69.1 | 84.8 (25.6) ** | −35.6 |
| ENNB | 0.9 | 72.2 (23.4) ** | 73.5 (12.7) * | 1.3 | 111 (45.9) ** | 38.8 |
| INTA | 1.3 | 60.2 (31.6) ** | 85.2 (77.6) ** | 25 | 38.5 (47.4) † | −21.7 |
| INTB | 2.5 | 64.7 (61.3) ** | -†† | - | 64.9 (70.1) ** | 0.2 |
| NIV | 7.5 | 77.3 (46.7) ** | 76.4 (46.7) ** | −0.9 | 109.4 (30.4) ** | 32.1 |
| STEG | 0.1 | 118.5 (20.1) ** | 135.6 (25.9) ** | 17.1 | 47.6 (28.5) † | −70.9 |
| VAL | 3.5 | 81.2 (83.2) ** | 33.8 (85.5) † | −47.4 | 33.2 (51) † | −48 |
| VERA | 1.3 | 116.3 (21.3) ** | 122.2 (13.7) * | 5.9 | 50 (53.9) † | −66.3 |
| ZEA | 25 | 83.9 (27) ** | 58.8 (29.2) † | −25.1 | 65.4 (50.8) ** | −18.5 |
| 3. Nine metabolites with unacceptable average recoveries from the initial experiment | ||||||
| AME | 2.6 | 26.1 (53.4) † | -†† | - | -†† | - |
| ASPH | 3.9 | 2.7 (53.2) † | 4.2 (80.5) † | 1.5 | 15.9 (77.2) † | 13.2 |
| CITRO | 42 | 33.8 (63.0) † | -†† | - | -†† | - |
| DON | 16 | 56.6 (15.6) † | 64.9 (35.8) ** | 8.3 | 108.7 (23.2) ** | 52.1 |
| ENNB1 | 1.7 | 41.4 (26.1) † | 52.2 (14.4) † | 10.8 | 67.5 (50) ** | 26.1 |
| EMOD | 0.7 | 7.7 (14.0) † | -†† | - | -†† | - |
| LOT | 25 | 162.3 (61.6) † | -†† | - | -†† | - |
| ROQC | 0.16 | 22 (48.3) † | 24.5 (75.2) † | 2.5 | 35.7 (73) † | 13.7 |
| USN | 16 | 10.7 (61.3) † | -†† | - | -†† | - |
| 4. No valid results obtained from the initial experiment | ||||||
| SKY | 62.5 | -†† | 53.3 (102.3) † | - | 53.8 (31.4) † | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rimayi, C.; Park, J.-H. Adjustment of Matrix Effects in Analysis of 36 Secondary Metabolites of Microbial and Plant Origin in Indoor Floor Dust Using Liquid Chromatography-Tandem Mass Spectrometry. Buildings 2023, 13, 1112. https://doi.org/10.3390/buildings13051112
Rimayi C, Park J-H. Adjustment of Matrix Effects in Analysis of 36 Secondary Metabolites of Microbial and Plant Origin in Indoor Floor Dust Using Liquid Chromatography-Tandem Mass Spectrometry. Buildings. 2023; 13(5):1112. https://doi.org/10.3390/buildings13051112
Chicago/Turabian StyleRimayi, Cornelius, and Ju-Hyeong Park. 2023. "Adjustment of Matrix Effects in Analysis of 36 Secondary Metabolites of Microbial and Plant Origin in Indoor Floor Dust Using Liquid Chromatography-Tandem Mass Spectrometry" Buildings 13, no. 5: 1112. https://doi.org/10.3390/buildings13051112