
Chemical Inhibitors Targeting the Histone Lysine Demethylase Families with Potential for Drug Discovery

Abstract
:1. Introduction
2. Inhibitors of FAD–Containing Lysine Demethylases


{kind=link}
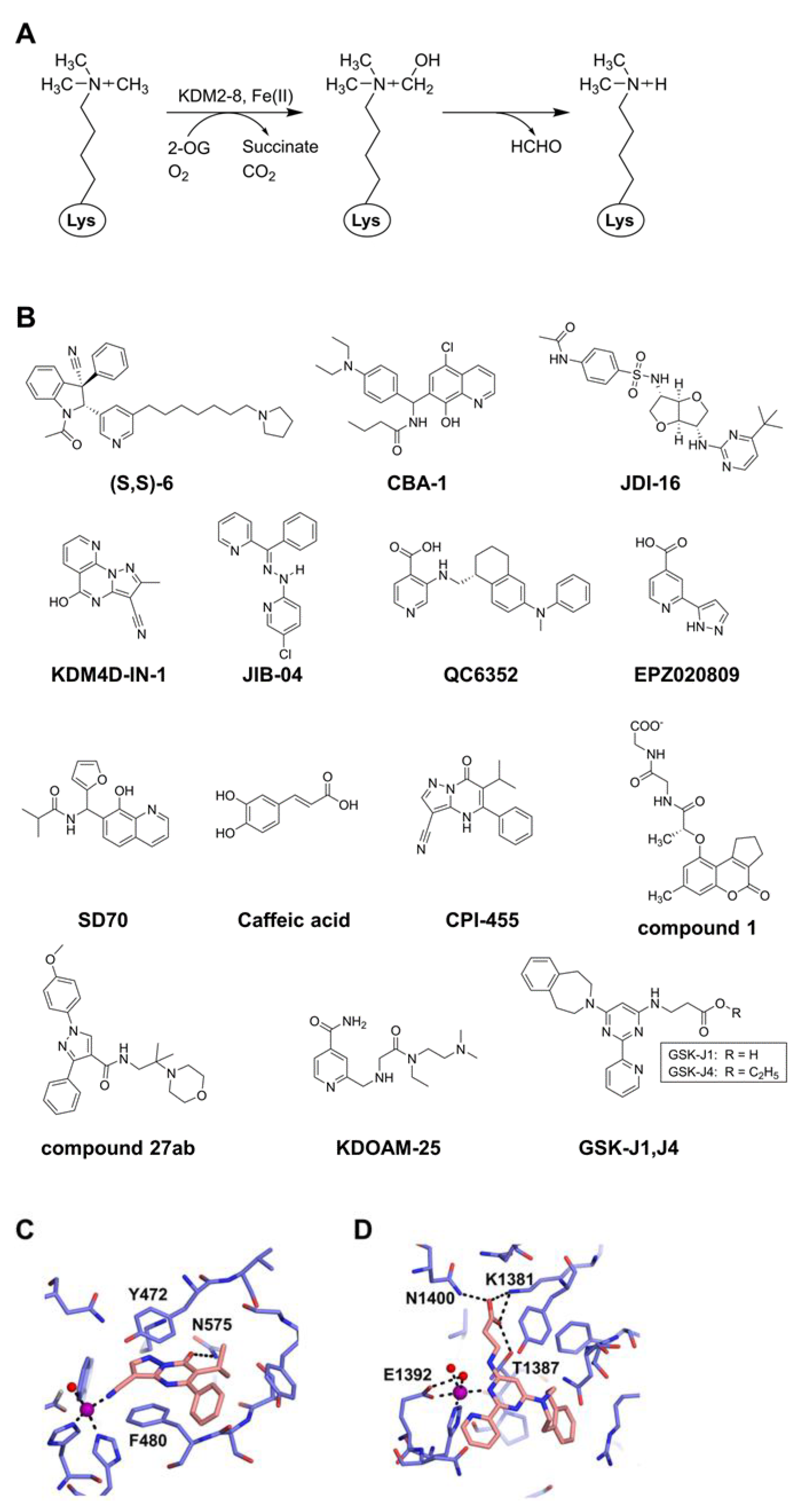{kind=link}
| Inhibitor | Target | Substrate | Potency | Application/Feature | Reference |
|---|---|---|---|---|---|
| ORY-1001 | KDM1A | H3K4me2 | 0.0086 μM 1,2 | Clinical trials for the treatment of AML (Phase Ib) and SCLC (Phase IIa) | [27] |
| S1024 | KDM1A/1B | H3K4me2 | 0.094 μM 1 | Chemical probe as a dual inhibitor of KDM1A and KDM1B for the study of H4K4me2 demethylation inhibition | [38] |
| IMG-7289 | KDM1A | H3K4me2 | 0.25 μM 1,2 | Clinical trials for the treatment of myeloid-related diseases (Phase II) and essential thrombocythemia (Phase IIb) | [39,40,41] |
| ORY-2001 | KDM1A | H3K4me2 | 0.10 μM 1 | Clinical trial (Phase IIa) for mild to moderate Alzheimer’s disease | [31,42] |
| CC-90011 | KDM1A | H3K4me2 | 0.017 μM 1,2 | Clinical trial (Phase I) for the treatment of neuroendocrine neoplasms and relapsed/refractory non-Hodgkin lymphoma | [28,43] |
3. Inhibitors of JmjC Domain–Containing Lysine Demethylases
3.1. Inhibitors of KDM2

3.2. Inhibitors of KDM3
3.3. Inhibitors of KDM4
3.4. Inhibitors of KDM5
3.5. Inhibitors of KDM6
| Inhibitor | Target | Substrate | Potency | Application | Reference |
|---|---|---|---|---|---|
| Compound (S,S)-6 | KDM2A, KDM7A | H3K36me2 | 0.16 μM 1 | Inhibits KDM2A-catalyzed demethylation in HeLa cells. | [63] |
| CBA-1 | KDM3A/3B | H3K9me2 | 3.9 μM 1 | Inhibits KDM3A overexpression in colon cancer cells and colon cancer organoids. | [72] |
| JDI-16 | KDM3C | H3K9 methylation | 0.82−6.12 μM 1 | Represses multiple KDM3C-dependent leukemia cell lines and patient-derived primary leukemic cells; shows substantial growth inhibitory abilities against multiple hematopoietic malignant cells. | [73] |
| KDM4D-IN-1 | KDM4D | H3K9 methylation | 0.41 μM 1 | Suppresses proliferation, induces apoptosis, and promotes angiogenesis of the renal cell carcinoma cells. | [82] |
| JIB-04 | KDM4A/4B/4C/4E, KDM6B | H3K9me3 | 5.0 μM 1 | Shows anti-cancer activity across several tumor types and in vivo mouse tumor xenografts; JIB-04 treatment induces cancer survival in an aggressive breast cancer model. | [87] |
| QC6352 | KDM4A/4B/4C/4D | H3K9me3, H3K36me3 | 35−104 nM (KDM4A−4D) 1 | Shows efficacy in patient-derived xenograft models of breast and colon cancers. | [89] |
| EPZ020809 | KDM4C | H3K9 methylation | 31 nM2 | No information available. | |
| TACH101 | A pan inhibitor of KDM4 | No information available | 0.004−0.072 µM (in gastric cancer cell lines) 1, 1–150 nM (in colorectal cancer cell lines) 1 | A Phase I clinical trial is ongoing for the treatment of gastrointestinal and high microsatellite instability metastatic colorectal cancers. | [90] |
| SD70 | KDM4C | H3K9me2 | 30 μM 1 | Inhibits the proliferation of prostate cancer cells and shows inhibition of tumor growth in vivo. | [92] |
| Caffeic acid | KDM4C | H3K9me2/me3 | 13.7 μM 1 | Effective against esophageal cancers; a Phase III clinical trial is ongoing for the treatment of esophageal squamous cell cancer; shows suppression of human glioma xenograft tumors. | [93,94,95,97] |
| CPI-455 | KDM5A/5B | H3K4me3 | 10 nM 1 | Attenuates the sphere formation of oral squamous cell carcinomas; effective against glioblastoma cells; effective against several KDM5-mediated drug-tolerant cancer cells such as HeLa, Colo829, and U2OS. | [64,105,106] |
| Cyclopenta[c] chromen derivative, compound 1 | KDM5A | H3K4me3 | 23.8 nM 1 | Shows efficacy against several KDM5A- overexpressing breast cancer cell lines such as MDA-MB-231, MCF-7, and MCF-10A. | [107] |
| Pyrazole derivative, compound 27 ab | KDM5B | H3K4me2/me3 | 0.0244 μM 1 | Inhibits proliferation and migration abilities of MKN45, a gastric cancer cell line | [108] |
| KDOAM-25 | KDM5A/5B/5C/5D | H3K4me3 | 71 nM (KDM5A), 19 nM (KDM5B), 69 nM (KDM5C and 5D) 1 | Impairs proliferation of multiple myeloma cell.s | [109] |
| GSK-J1/J4 | KDM6A/6B | H3K27me2/me3 | 60 nM1 | Shows antitumor efficacy in several cancers, such as glioma and leukemia; effective to reduce tumor volume in mice xenograft models; suppresses KDM6B-mediated proinflammatory responses in macrophages. | [118,119,120,121,122,123] |
| Caffeic acid | KDM6A | Not studied | 5.5 μM 1 | No information available. | [94] |
3.6. Inhibitors of KDM7
3.7. Inhibitors of KDM8
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ptashne, M. Principles of a switch. Nat. Chem. Biol. 2011, 7, 484–487. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornberg, R.D.; Lorch, Y. Twenty-Five Years of the Nucleosome, Fundamental Particle of the Eukaryote Chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Baarends, W.M.; Hoogerbrugge, J.W.; Roest, H.P.; Ooms, M.; Vreeburg, J.; Hoeijmakers, J.H.; Grootegoed, J.A. Histone Ubiquitination and Chromatin Remodeling in Mouse Spermatogenesis. Dev. Biol. 1999, 207, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Attikum, H.; Gasser, S.M. The histone code at DNA breaks: A guide to repair? Nat. Rev. Mol. Cell Biol. 2005, 6, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, J.; Kellis, M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010, 28, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hojfeldt, J.W.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013, 12, 917–930. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Karytinos, A.; Forneris, F.; Profumo, A.; Ciossani, G.; Battaglioli, E.; Binda, C.; Mattevi, A. A Novel Mammalian Flavin-dependent Histone Demethylase. J. Biol. Chem. 2009, 284, 17775–17782. [Google Scholar] [CrossRef] [Green Version]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef]
- Bhat, K.P.; Umit Kaniskan, H.; Jin, J.; Gozani, O. Epigenetics and beyond: Targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discov. 2021, 20, 265–286. [Google Scholar] [CrossRef]
- Jambhekar, A.; Anastas, J.N.; Shi, Y. Histone Lysine Demethylase Inhibitors. Cold Spring Harb. Perspect. Med. 2017, 7, a026484. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef] [PubMed]
- D’Oto, A.; Tian, Q.-W.; Davidoff, A.M.; Yang, J. Histone Demethylases and Their Roles in Cancer Epigenetics. J. Med. Oncol. Ther. 2016, 1, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manni, W.; Jianxin, X.; Weiqi, H.; Siyuan, C.; Huashan, S. JMJD family proteins in cancer and inflammation. Signal Transduct. Target. Ther. 2022, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H.; Umehara, T. Structural insight into inhibitors of flavin adenine dinucleotide-dependent lysine demethylases. Epigenetics 2017, 12, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, P.F. Oxidation of amines by flavoproteins. Arch. Biochem. Biophys. 2010, 493, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Culhane, J.C.; Cole, P.A. LSD1 and the chemistry of histone demethylation. Curr. Opin. Chem. Biol. 2007, 11, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018, 33, 495–511.e12. [Google Scholar] [CrossRef] [Green Version]
- Kanouni, T.; Severin, C.; Cho, R.W.; Yuen, N.Y.-Y.; Xu, J.; Shi, L.; Lai, C.; Del Rosario, J.R.; Stansfield, R.K.; Lawton, L.N.; et al. Discovery of CC-90011: A Potent and Selective Reversible Inhibitor of Lysine Specific Demethylase 1 (LSD1). J. Med. Chem. 2020, 63, 14522–14529. [Google Scholar] [CrossRef]
- Dai, X.-J.; Liu, Y.; Xiong, X.-P.; Xue, L.-P.; Zheng, Y.-C.; Liu, H.-M. Tranylcypromine Based Lysine-Specific Demethylase 1 Inhibitor: Summary and Perspective. J. Med. Chem. 2020, 63, 14197–14215. [Google Scholar] [CrossRef]
- Dai, X.-J.; Liu, Y.; Xue, L.-P.; Xiong, X.-P.; Zhou, Y.; Zheng, Y.-C.; Liu, H.-M. Reversible Lysine Specific Demethylase 1 (LSD1) Inhibitors: A Promising Wrench to Impair LSD1. J. Med. Chem. 2021, 64, 2466–2488. [Google Scholar] [CrossRef]
- Fang, Y.; Liao, G.; Yu, B. LSD1/KDM1A inhibitors in clinical trials: Advances and prospects. J. Hematol. Oncol. 2019, 12, 129. [Google Scholar] [CrossRef] [Green Version]
- Mehndiratta, S.; Liou, J.-P. Histone lysine specific demethylase 1 inhibitors. RSC Med. Chem. 2020, 11, 969–981. [Google Scholar] [CrossRef]
- Anan, K.; Hino, S.; Shimizu, N.; Sakamoto, A.; Nagaoka, K.; Takase, R.; Kohrogi, K.; Araki, H.; Hino, Y.; Usuki, S.; et al. LSD1 mediates metabolic reprogramming by glucocorticoids during myogenic differentiation. Nucleic Acids Res. 2018, 46, 5441–5454. [Google Scholar] [CrossRef] [Green Version]
- Hino, S.; Sakamoto, A.; Nagaoka, K.; Anan, K.; Wang, Y.; Mimasu, S.; Umehara, T.; Yokoyama, S.; Kosai, K.-I.; Nakao, M. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun. 2012, 3, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimasu, S.; Umezawa, N.; Sato, S.; Higuchi, T.; Umehara, T.; Yokoyama, S. Structurally Designed trans-2-Phenylcyclopropylamine Derivatives Potently Inhibit Histone Demethylase LSD1/KDM1. Biochemistry 2010, 49, 6494–6503. [Google Scholar] [CrossRef]
- Ramms, B.; Pollow, D.P.; Zhu, H.; Nora, C.; Harrington, A.R.; Omar, I.; Gordts, P.L.; Wortham, M.; Sander, M. Systemic LSD1 Inhibition Prevents Aberrant Remodeling of Metabolism in Obesity. Diabetes 2022, 71, 2513–2529. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Valente, S.; Romanenghi, M.; Pilotto, S.; Cirilli, R.; Karytinos, A.; Ciossani, G.; Botrugno, O.A.; Forneris, F.; Tardugno, M.; et al. Biochemical, Structural, and Biological Evaluation of Tranylcypromine Derivatives as Inhibitors of Histone Demethylases LSD1 and LSD2. J. Am. Chem. Soc. 2010, 132, 6827–6833. [Google Scholar] [CrossRef]
- Niwa, H.; Watanabe, C.; Sato, S.; Harada, T.; Watanabe, H.; Tabusa, R.; Fukasawa, S.; Shiobara, A.; Hashimoto, T.; Ohno, O.; et al. Structure–Activity Relationship and In Silico Evaluation of cis- and trans-PCPA-Derived Inhibitors of LSD1 and LSD2. ACS Med. Chem. Lett. 2022, 13, 1485–1492. [Google Scholar] [CrossRef]
- Jutzi, J.S.; Kleppe, M.; Dias, J.; Staehle, H.F.; Shank, K.; Teruya-Feldstein, J.; Gambheer, S.M.M.; Dierks, C.; Rienhoff, H.Y., Jr.; Levine, R.L.; et al. LSD1 Inhibition Prolongs Survival in Mouse Models of MPN by Selectively Targeting the Disease Clone. Hemasphere 2018, 2, e54. [Google Scholar] [CrossRef] [PubMed]
- Gill, H. Lysine-Specific Demethylase 1 (LSD1/KDM1A) Inhibition as a Target for Disease Modification in Myelofibrosis. Cells 2022, 11, 2107. [Google Scholar] [CrossRef]
- Palandri, F.; Vianelli, N.; Ross, D.M.; Cochrane, T.; Lane, S.W.; Larsen, S.R.; Gerds, A.T.; Halpern, A.B.; Shortt, J.; Rossetti, J.M.; et al. A Phase 2 Study of the LSD1 Inhibitor Img-7289 (bomedemstat) for the Treatment of Essential Thrombocythemia (ET). Blood 2021, 138, 386. [Google Scholar] [CrossRef]
- Maes, T.; Mascaró, C.; Rotllant, D.; Lufino, M.M.P.; Estiarte, A.; Guibourt, N.; Cavalcanti, F.; Griñan-Ferré, C.; Pallàs, M.; Nadal, R.; et al. Modulation of KDM1A with vafidemstat rescues memory deficit and behavioral alterations. PLoS ONE 2020, 15, e0233468. [Google Scholar] [CrossRef]
- Hollebecque, A.; Salvagni, S.; Plummer, R.; Niccoli, P.; Capdevila, J.; Curigliano, G.; Moreno, V.; de Braud, F.; de Villambrosia, S.G.; Martin-Romano, P.; et al. Clinical activity of CC-90011, an oral, potent, and reversible LSD1 inhibitor, in advanced malignancies. Cancer 2022, 128, 3185–3195. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.-I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Maiques-Diaz, A.; Spencer, G.J.; Lynch, J.T.; Ciceri, F.; Williams, E.L.; Amaral, F.M.R.; Wiseman, D.H.; Harris, W.J.; Li, Y.; Sahoo, S.; et al. Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia. Cell Rep. 2018, 22, 3641–3659. [Google Scholar] [CrossRef] [Green Version]
- Saleque, S.; Kim, J.; Rooke, H.M.; Orkin, S.H. Epigenetic Regulation of Hematopoietic Differentiation by Gfi-1 and Gfi-1b Is Mediated by the Cofactors CoREST and LSD1. Mol. Cell 2007, 27, 562–572. [Google Scholar] [CrossRef]
- Takagi, S.; Ishikawa, Y.; Mizutani, A.; Iwasaki, S.; Matsumoto, S.; Kamada, Y.; Nomura, T.; Nakamura, K. LSD1 Inhibitor T-3775440 Inhibits SCLC Cell Proliferation by Disrupting LSD1 Interactions with SNAG Domain Proteins INSM1 and GFI1B. Cancer Res 2017, 77, 4652–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinyard, M.E.; Su, C.; Siegenfeld, A.P.; Waterbury, A.L.; Freedy, A.M.; Gosavi, P.M.; Park, Y.; Kwan, E.E.; Senzer, B.; Doench, J.G.; et al. CRISPR-suppressor scanning reveals a nonenzymatic role of LSD1 in AML. Nat. Chem. Biol. 2019, 15, 529–539. [Google Scholar] [CrossRef]
- Hattori, Y.; Matsumoto, S.; Morimoto, S.; Daini, M.; Toyofuku, M.; Matsuda, S.; Baba, R.; Murakami, K.; Iwatani, M.; Oki, H.; et al. Design, synthesis, and structure–activity relationship of TAK-418 and its derivatives as a novel series of LSD1 inhibitors with lowered risk of hematological side effects. Eur. J. Med. Chem. 2022, 239, 114522. [Google Scholar] [CrossRef]
- Sacilotto, N.; Dessanti, P.; Lufino, M.M.P.; Ortega, A.; Rodríguez-Gimeno, A.; Salas, J.; Maes, T.; Buesa, C.; Mascaró, C.; Soliva, R. Comprehensive in Vitro Characterization of the LSD1 Small Molecule Inhibitor Class in Oncology. ACS Pharmacol. Transl. Sci. 2021, 4, 1818–1834. [Google Scholar] [CrossRef]
- Klose, R.J.; Yamane, K.; Bae, Y.; Zhang, D.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 2006, 442, 312–316. [Google Scholar] [CrossRef]
- Tsukada, Y.-I.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- McDonough, M.A.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672. [Google Scholar] [CrossRef]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef]
- Yang, J.; Hu, Y.; Zhang, B.; Liang, X.; Li, X. The JMJD Family Histone Demethylases in Crosstalk Between Inflammation and Cancer. Front. Immunol. 2022, 13, 881396. [Google Scholar] [CrossRef]
- He, X.; Zhang, H.; Zhang, Y.; Ye, Y.; Wang, S.; Bai, R.; Xie, T.; Ye, X.-Y. Drug discovery of histone lysine demethylases (KDMs) inhibitors (progress from 2018 to present). Eur. J. Med. Chem. 2022, 231, 114143. [Google Scholar] [CrossRef]
- McAllister, T.E.; England, K.S.; Hopkinson, R.J.; Brennan, P.E.; Kawamura, A.; Schofield, C.J. Recent Progress in Histone Demethylase Inhibitors. J. Med. Chem. 2016, 59, 1308–1329. [Google Scholar] [CrossRef] [PubMed]
- Tzatsos, A.; Pfau, R.; Kampranis, S.C.; Tsichlis, P.N. Ndy1/KDM2B immortalizes mouse embryonic fibroblasts by repressing the Ink4a / Arf locus. Proc. Natl. Acad. Sci. USA 2009, 106, 2641–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfau, R.; Tzatsos, A.; Kampranis, S.C.; Serebrennikova, O.B.; Bear, S.E.; Tsichlis, P.N. Members of a family of JmjC domain-containing oncoproteins immortalize embryonic fibroblasts via a JmjC domain-dependent process. Proc. Natl. Acad. Sci. USA 2008, 105, 1907–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Kallin, E.M.; Tsukada, Y.-I.; Zhang, Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15Ink4b. Nat. Struct. Mol. Biol. 2008, 15, 1169–1175. [Google Scholar] [CrossRef]
- Tzatsos, A.; Paskaleva, P.; Ferrari, F.; Deshpande, V.; Stoykova, S.; Contino, G.; Wong, K.-K.; Lan, F.; Trojer, P.; Park, P.J.; et al. KDM2B promotes pancreatic cancer via Polycomb-dependent and -independent transcriptional programs. J. Clin. Investig. 2013, 123, 727–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Yang, X.; Wang, H.; Shao, Q. The critical role of histone lysine demethylase KDM2B in cancer. Am. J. Transl. Res. 2018, 10, 2222–2233. [Google Scholar] [PubMed]
- Gerken, P.A.; Wolstenhulme, J.R.; Tumber, A.; Hatch, S.B.; Zhang, Y.; Müller, S.; Chandler, S.A.; Mair, B.; Li, F.; Nijman, S.M.B.; et al. Discovery of a Highly Selective Cell-Active Inhibitor of the Histone Lysine Demethylases KDM2/7. Angew. Chem. Int. Ed. 2017, 56, 15555–15559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; Tindell, C.A.; Wilson, C.; Williamson, K.E.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef]
- Kruidenier, L.; Chung, C.-W.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408. [Google Scholar] [CrossRef] [Green Version]
- Yamane, K.; Toumazou, C.; Tsukada, Y.-I.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-Containing H3K9 Demethylase, Facilitates Transcription Activation by Androgen Receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhou, S.; Liao, L.; Chen, X.; Meistrich, M.; Xu, J. Jmjd1a Demethylase-regulated Histone Modification Is Essential for cAMP-response Element Modulator-regulated Gene Expression and Spermatogenesis. J. Biol. Chem. 2010, 285, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Scott, G.; Ray, M.K.; Mishina, Y.; Zhang, Y. Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature 2007, 450, 119–123. [Google Scholar] [CrossRef]
- Krieg, A.J.; Rankin, E.B.; Chan, D.; Razorenova, O.; Fernandez, S.; Giaccia, A.J. Regulation of the Histone Demethylase JMJD1A by Hypoxia-Inducible Factor 1α Enhances Hypoxic Gene Expression and Tumor Growth. Mol. Cell. Biol. 2010, 30, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, S.; Kitadate, A.; Abe, F.; Takahashi, N.; Tagawa, H. Hypoxia-inducible KDM3A addiction in multiple myeloma. Blood Adv. 2018, 2, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.; Su, G.; Ji, J.; Yang, X.; Miao, M.; Mo, P.; Li, M.; Xu, J.; Li, W.; Yu, C. Histone demethylase JMJD1A promotes colorectal cancer growth and metastasis by enhancing Wnt/β-catenin signaling. J. Biol. Chem. 2018, 293, 10606–10619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Sviripa, V.M.; Xie, Y.; Yu, T.; Haney, M.G.; Blackburn, J.S.; Adeniran, C.A.; Zhan, C.-G.; Watt, D.S.; Liu, C. Epigenetic Regulation of Wnt Signaling by Carboxamide-Substituted Benzhydryl Amines that Function as Histone Demethylase Inhibitors. iScience 2020, 23, 101795. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, L.; Hu, L.; Dirks, W.G.; Zhao, Y.; Wei, Z.; Chen, D.; Li, Z.; Wang, Z.; Han, Y.; et al. Small molecular modulators of JMJD1C preferentially inhibit growth of leukemia cells. Int. J. Cancer 2020, 146, 400–412. [Google Scholar] [CrossRef]
- Cloos, P.A.C.; Christensen, J.; Agger, K.; Maiolica, A.; Rappsilber, J.; Antal, T.; Hansen, K.H.; Helin, K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006, 442, 307–311. [Google Scholar] [CrossRef]
- Fodor, B.D.; Kubicek, S.; Yonezawa, M.; O’Sullivan, R.J.; Sengupta, R.; Perez-Burgos, L.; Opravil, S.; Mechtler, K.; Schotta, G.; Jenuwein, T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006, 20, 1557–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of Histone Lysine Trimethylation by the JMJD2 Family of Histone Demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.C.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef]
- Liu, G.; Bollig-Fischer, A.; Kreike, B.; van de Vijver, M.J.; Abrams, J.; Ethier, S.P.; Yang, Z.Q. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene 2009, 28, 4491–4500. [Google Scholar] [CrossRef] [Green Version]
- Rui, L.; Emre, N.T.; Kruhlak, M.J.; Chung, H.-J.; Steidl, C.; Slack, G.; Wright, G.W.; Lenz, G.; Ngo, V.N.; Shaffer, A.L.; et al. Cooperative Epigenetic Modulation by Cancer Amplicon Genes. Cancer Cell 2010, 18, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Loh, Y.-H.; Zhang, W.; Chen, X.; George, J.; Ng, H.-H. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007, 21, 2545–2557. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Young, B.; Wang, Y.; Davidoff, A.M.; Rankovic, Z.; Yang, J. Recent Advances with KDM4 Inhibitors and Potential Applications. J. Med. Chem. 2022, 65, 9564–9579. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhu, L.; Zhang, J.; Lin, Z. Histone demethylase KDM4D inhibition suppresses renal cancer progression and angiogenesis through JAG1 signaling. Cell Death Discov. 2021, 7, 284. [Google Scholar] [CrossRef]
- King, O.; Li, X.S.; Sakurai, M.; Kawamura, A.; Rose, N.R.; Ng, S.S.; Quinn, A.M.; Rai, G.; Mott, B.T.; Beswick, P.; et al. Quantitative High-Throughput Screening Identifies 8-Hydroxyquinolines as Cell-Active Histone Demethylase Inhibitors. PLoS ONE 2010, 5, e15535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Li, M.; Zhuo, M.; Guo, P.; Chen, Q.; Mo, P.; Li, W.; Yu, C. Histone demethylase JMJD2D promotes the self-renewal of liver cancer stem-like cells by enhancing EpCAM and Sox9 expression. J. Biol. Chem. 2021, 296, 100121. [Google Scholar] [CrossRef]
- Peng, K.; Zhuo, M.; Li, M.; Chen, Q.; Mo, P.; Yu, C. Histone demethylase JMJD2D activates HIF1 signaling pathway via multiple mechanisms to promote colorectal cancer glycolysis and progression. Oncogene 2020, 39, 7076–7091. [Google Scholar] [CrossRef]
- Zhuo, M.; Chen, W.; Shang, S.; Guo, P.; Peng, K.; Li, M.; Mo, P.; Zhang, Y.; Qiu, X.; Li, W.; et al. Inflammation-induced JMJD2D promotes colitis recovery and colon tumorigenesis by activating Hedgehog signaling. Oncogene 2020, 39, 3336–3353. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A.M.; Ruiz, J.; Bruick, R.; Peña-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 2035. [Google Scholar] [CrossRef] [Green Version]
- Cascella, B.; Lee, S.G.; Singh, S.; Jez, J.M.; Mirica, L.M. The small molecule JIB-04 disrupts O2binding in the Fe-dependent histone demethylase KDM4A/JMJD2A. Chem. Commun. 2017, 53, 2174–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.K.; Bonaldi, T.; Cuomo, A.; Del Rosario, J.R.; Hosfield, D.J.; Kanouni, T.; Kao, S.-C.; Lai, C.; Lobo, N.A.; Matuszkiewicz, J.; et al. Design of KDM4 Inhibitors with Antiproliferative Effects in Cancer Models. ACS Med. Chem. Lett. 2017, 8, 869–874. [Google Scholar] [CrossRef]
- Perabo, F.; Chandhasin, C.; Yoo, S.; Dang, V.; Del Rosario, J.; Chen, Y.K.; Stafford, J.; Quake, S.; Clarke, M.F. TACH101, a first-in-class pan-inhibitor of KDM4 for treatment of gastrointestinal cancers. J. Clin. Oncol. 2022, 40, 132. [Google Scholar] [CrossRef]
- Wigle, T.J.; Swinger, K.K.; Campbell, J.E.; Scholle, M.D.; Sherrill, J.; Admirand, E.A.; Boriack-Sjodin, P.A.; Kuntz, K.W.; Chesworth, R.; Moyer, M.P.; et al. A High-Throughput Mass Spectrometry Assay Coupled with Redox Activity Testing Reduces Artifacts and False Positives in Lysine Demethylase Screening. J. Biomol. Screen. 2015, 20, 810–820. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Yang, L.; Xie, M.; Lin, C.; Merkurjev, D.; Yang, J.C.; Tanasa, B.; Oh, S.; Zhang, J.; Ohgi, K.A.; et al. Chem-seq permits identification of genomic targets of drugs against androgen receptor regulation selected by functional phenotypic screens. Proc. Natl. Acad. Sci. USA 2014, 111, 9235–9240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, R.; Mi, Y.; Yuan, X.; Kong, D.; Li, W.; Li, R.; Wang, B.; Zhu, Y.; Kong, J.; Ma, Z.; et al. GASC1-Adapted Neoadjuvant Chemotherapy for Resectable Esophageal Squamous Cell Carcinoma: A Prospective Clinical Biomarker Trial. J. Oncol. 2020, 2020, 1607860. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.L.; Kristensen, L.H.; Stephansen, K.B.; Kristensen, J.B.; Helgstrand, C.; Lees, M.; Cloos, P.; Helin, K.; Gajhede, M.; Olsen, L. Identification of catechols as histone-lysine demethylase inhibitors. FEBS Lett. 2012, 586, 1190–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, R.; Yang, L.; Yuan, X.; Kong, J.; Liu, Y.; Yin, W.; Gao, S.; Zhang, Y. GASC1 Promotes Stemness of Esophageal Squamous Cell Carcinoma via NOTCH1 Promoter Demethylation. J. Oncol. 2019, 2019, 1621054. [Google Scholar] [CrossRef] [PubMed]
- Olthof, M.R.; Hollman, P.C.H.; Katan, M.B. Chlorogenic Acid and Caffeic Acid Are Absorbed in Humans. J. Nutr. 2001, 131, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.; Yang, X.; Liu, Z.; Shao, Z.; Song, C.; Zhang, K.; Wang, X.; Li, Z. GASC1 promotes glioma progression by enhancing NOTCH1 signaling. Mol. Med. Rep. 2021, 23, 1–14. [Google Scholar] [CrossRef]
- Christensen, J.; Agger, K.; Cloos, P.A.; Pasini, D.; Rose, S.; Sennels, L.; Rappsilber, J.; Hansen, K.H.; Salcini, A.E.; Helin, K. RBP2 Belongs to a Family of Demethylases, Specific for Tri-and Dimethylated Lysine 4 on Histone 3. Cell 2007, 128, 1063–1076. [Google Scholar] [CrossRef] [Green Version]
- Iwase, S.; Lan, F.; Bayliss, P.; De La Torre-Ubieta, L.; Huarte, M.; Qi, H.H.; Whetstine, J.R.; Bonni, A.; Roberts, T.M.; Shi, Y. The X-Linked Mental Retardation Gene SMCX/JARID1C Defines a Family of Histone H3 Lysine 4 Demethylases. Cell 2007, 128, 1077–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamane, K.; Tateishi, K.; Klose, R.J.; Fang, J.; Fabrizio, L.A.; Erdjument-Bromage, H.; Taylor-Papadimitriou, J.; Tempst, P.; Zhang, Y. PLU-1 Is an H3K4 Demethylase Involved in Transcriptional Repression and Breast Cancer Cell Proliferation. Mol. Cell 2007, 25, 801–812. [Google Scholar] [CrossRef]
- Barrett, A.; Madsen, B.; Copier, J.; Lu, P.J.; Cooper, L.; Scibetta, A.G.; Burchell, J.; Taylor-Papadimitriou, J. PLU-1 nuclear protein, which is upregulated in breast cancer, shows restricted expression in normal human adult tissues: A new cancer/testis antigen? Int. J. Cancer 2002, 101, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Hayami, S.; Yoshimatsu, M.; Veerakumarasivam, A.; Unoki, M.; Iwai, Y.; Tsunoda, T.; Field, H.I.; Kelly, J.D.; Neal, D.E.; Yamaue, H.; et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: Involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol. Cancer 2010, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A Temporarily Distinct Subpopulation of Slow-Cycling Melanoma Cells Is Required for Continuous Tumor Growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahiliani, M.; Mei, P.; Fang, R.; Leonor, T.; Rutenberg, M.; Shimizu, F.; Li, J.; Rao, A.; Shi, Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature 2007, 447, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Facompre, N.D.; Harmeyer, K.M.; Sahu, V.; Gimotty, P.A.; Rustgi, A.K.; Nakagawa, H.; Basu, D. Targeting JARID1B’s demethylase activity blocks a subset of its functions in oral cancer. Oncotarget 2018, 9, 8985–8998. [Google Scholar] [CrossRef] [Green Version]
- Banelli, B.; Carra, E.; Barbieri, F.; Würth, R.; Parodi, F.; Pattarozzi, A.; Carosio, R.; Forlani, A.; Allemanni, G.; Marubbi, D.; et al. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma. Cell Cycle 2015, 14, 3418–3429. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.-J.; Ko, C.-N.; Zhong, H.-J.; Leung, C.-H.; Ma, D.-L. Structure-Based Discovery of a Selective KDM5A Inhibitor that Exhibits Anti-Cancer Activity via Inducing Cell Cycle Arrest and Senescence in Breast Cancer Cell Lines. Cancers 2019, 11, 92. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Liang, Q.; Ren, H.; Zhang, X.; Wu, Y.; Zhang, K.; Ma, L.-Y.; Zheng, Y.-C.; Liu, H.-M. Discovery of pyrazole derivatives as cellular active inhibitors of histone lysine specific demethylase 5B (KDM5B/JARID1B). Eur. J. Med. Chem. 2020, 192, 112161. [Google Scholar] [CrossRef]
- Tumber, A.; Nuzzi, A.; Hookway, E.S.; Hatch, S.B.; Velupillai, S.; Johansson, C.; Kawamura, A.; Savitsky, P.; Yapp, C.; Szykowska, A.; et al. Potent and Selective KDM5 Inhibitor Stops Cellular Demethylation of H3K4me3 at Transcription Start Sites and Proliferation of MM1S Myeloma Cells. Cell Chem. Biol. 2017, 24, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankowska, A.M.; Makishima, H.; Tiu, R.V.; Szpurka, H.; Huang, Y.; Traina, F.; Visconte, V.; Sugimoto, Y.; Prince, C.; O’Keefe, C.; et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood 2011, 118, 3932–3941. [Google Scholar] [CrossRef] [Green Version]
- Patani, N.; Jiang, W.G.; Newbold, R.F.; Mokbel, K. Histone-modifier gene expression profiles are associated with pathological and clinical outcomes in human breast cancer. Anticancer. Res. 2011, 31, 4115–4125. [Google Scholar]
- Agger, K.; Cloos, P.A.; Rudkjær, L.; Williams, K.; Andersen, G.; Christensen, J.; Helin, K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A–ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009, 23, 1171–1176. [Google Scholar] [CrossRef] [Green Version]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenführ, M.; Maertens, G.; Banck, M.; Zhou, M.-M.; Walsh, M.J.; et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderton, J.A.; Bose, S.; Vockerodt, M.; Vrzalikova, K.; Wei, W.; Kuo, M.; Helin, K.; Christensen, J.G.; Rowe, M.; Murray, P.G.; et al. The H3K27me3 demethylase, KDM6B, is induced by Epstein–Barr virus and over-expressed in Hodgkin’s Lymphoma. Oncogene 2011, 30, 2037–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The Histone H3 Lysine-27 Demethylase Jmjd3 Links Inflammation to Inhibition of Polycomb-Mediated Gene Silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, M.; Sheng, M.; Zhang, P.; Chen, Z.; Xing, W.; Bai, J.; Cheng, T.; Yang, F.-C.; Zhou, Y. Therapeutic potential of GSK-J4, a histone demethylase KDM6B/JMJD3 inhibitor, for acute myeloid leukemia. J. Cancer Res. Clin. Oncol. 2018, 144, 1065–1077. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, A.; Fiveash, J.B.; Yang, E.S. Combined Targeting of Mutant p53 and Jumonji Family Histone Demethylase Augments Therapeutic Efficacy of Radiation in H3K27M DIPG. Int. J. Mol. Sci. 2020, 21, 490. [Google Scholar] [CrossRef] [Green Version]
- Illiano, M.; Conte, M.; Salzillo, A.; Ragone, A.; Spina, A.; Nebbioso, A.; Altucci, L.; Sapio, L.; Naviglio, S. The KDM Inhibitor GSKJ4 Triggers CREB Downregulation via a Protein Kinase A and Proteasome-Dependent Mechanism in Human Acute Myeloid Leukemia Cells. Front. Oncol. 2020, 10, 799. [Google Scholar] [CrossRef]
- Horton, J.R.; Engstrom, A.; Zoeller, E.L.; Liu, X.; Shanks, J.R.; Zhang, X.; Johns, M.A.; Vertino, P.M.; Fu, H.; Cheng, X. Characterization of a Linked Jumonji Domain of the KDM5/JARID1 Family of Histone H3 Lysine 4 Demethylases. J. Biol. Chem. 2016, 291, 2631–2646. [Google Scholar] [CrossRef] [Green Version]
- Sakaki, H.; Okada, M.; Kuramoto, K.; Takeda, H.; Watarai, H.; Suzuki, S.; Seino, S.; Seino, M.; Ohta, T.; Nagase, S.; et al. GSKJ4, A Selective Jumonji H3K27 Demethylase Inhibitor, Effectively Targets Ovarian Cancer Stem Cells. Anticancer. Res. 2015, 35, 6607–6614. [Google Scholar] [PubMed]
- Benyoucef, A.; Palii, C.G.; Wang, C.; Porter, C.J.; Chu, A.; Dai, F.; Tremblay, V.; Rakopoulos, P.; Singh, K.; Huang, S.; et al. UTX inhibition as selective epigenetic therapy against TAL1-driven T-cell acute lymphoblastic leukemia. Genes Dev. 2016, 30, 508–521. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.R.; Upadhyay, A.K.; Qi, H.H.; Zhang, X.; Shi, Y.; Cheng, X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 2010, 17, 38–43. [Google Scholar] [CrossRef]
- Tsukada, Y.-I.; Ishitani, T.; Nakayama, K.I. KDM7 is a dual demethylase for histone H3 Lys 9 and Lys 27 and functions in brain development. Genes Dev. 2010, 24, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Li, J.; Song, T.; Lu, M.; Kan, P.-Y.; Lee, M.G.; Sha, B.; Shi, X. Recognition of Histone H3K4 Trimethylation by the Plant Homeodomain of PHF2 Modulates Histone Demethylation. J. Biol. Chem. 2010, 285, 9322–9326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleine-Kohlbrecher, D.; Christensen, J.; Vandamme, J.; Abarrategui, I.; Bak, M.; Tommerup, N.; Shi, X.; Gozani, O.; Rappsilber, J.; Salcini, A.E.; et al. A Functional Link between the Histone Demethylase PHF8 and the Transcription Factor ZNF711 in X-Linked Mental Retardation. Mol. Cell 2010, 38, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Xiang, Y.; Wang, Y.; Li, X.; Xu, L.; Zhu, Z.; Zhang, T.; Zhu, Q.; Zhang, K.; Jing, N.; et al. Dual-specificity histone demethylase KIAA1718 (KDM7A) regulates neural differentiation through FGF4. Cell Res. 2010, 20, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 Is a Histone Arginine Demethylase. Science 2007, 318, 444–447. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, C.; Takahashi, K.; Hayama, S.; Ishikawa, N.; Kato, T.; Ito, T.; Tsuchiya, E.; Nakamura, Y.; Daigo, Y. Identification of Myc-associated protein with JmjC domain as a novel therapeutic target oncogene for lung cancer. Mol. Cancer Ther. 2007, 6, 542–551. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Tie, Y.; Fang, Z.; Wu, X.; Yi, T.; Huang, S.; Liang, X.; Qian, Y.; Wang, X.; Pi, R.; et al. Jumonji domain-containing 6 (JMJD6) identified as a potential therapeutic target in ovarian cancer. Signal Transduct. Target. Ther. 2019, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhang, R.; Liu, Y.; Fang, Z.; Zhang, H.; Fan, Y.; Yang, S.; Xiang, R. Discovery of a new class of JMJD6 inhibitors and structure–activity relationship study. Bioorganic Med. Chem. Lett. 2021, 44, 128109. [Google Scholar] [CrossRef] [PubMed]
- Baby, S.; Gurukkala Valapil, D.; Shankaraiah, N. Unravelling KDM4 histone demethylase inhibitors for cancer therapy. Drug Discov. Today 2021, 26, 1841–1856. [Google Scholar] [CrossRef] [PubMed]
- Gilham, D.; Wasiak, S.; Tsujikawa, L.M.; Halliday, C.; Norek, K.; Patel, R.G.; Kulikowski, E.; Johansson, J.; Sweeney, M.; Wong, N.C. RVX-208, a BET-inhibitor for treating atherosclerotic cardiovascular disease, raises ApoA-I/HDL and represses pathways that contribute to cardiovascular disease. Atherosclerosis 2016, 247, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Moreno, J.M.; Fontecha-Barriuso, M.; Martin-Sanchez, D.; Guerrero-Mauvecin, J.; Goma-Garces, E.; Fernandez-Fernandez, B.; Carriazo, S.; Sanchez-Niño, M.D.; Ramos, A.M.; Ruiz-Ortega, M.; et al. Epigenetic Modifiers as Potential Therapeutic Targets in Diabetic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 4113. [Google Scholar] [CrossRef] [PubMed]
- Neele, A.E.; Willemsen, L.; Chen, H.-J.; Dzobo, K.E.; De Winther, M.P. Targeting epigenetics as atherosclerosis treatment: An updated view. Curr. Opin. Lipidol. 2020, 31, 324–330. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, N.D.; Niwa, H.; Umehara, T. Chemical Inhibitors Targeting the Histone Lysine Demethylase Families with Potential for Drug Discovery. Epigenomes 2023, 7, 7. https://doi.org/10.3390/epigenomes7010007
Das ND, Niwa H, Umehara T. Chemical Inhibitors Targeting the Histone Lysine Demethylase Families with Potential for Drug Discovery. Epigenomes. 2023; 7(1):7. https://doi.org/10.3390/epigenomes7010007
Chicago/Turabian StyleDas, Nando Dulal, Hideaki Niwa, and Takashi Umehara. 2023. "Chemical Inhibitors Targeting the Histone Lysine Demethylase Families with Potential for Drug Discovery" Epigenomes 7, no. 1: 7. https://doi.org/10.3390/epigenomes7010007