
Systematic Functional Annotation Workflow for Insects



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Programs Comprising the Workflow
2.2. Insects and Sample Collection
2.3. RNA Sequencing
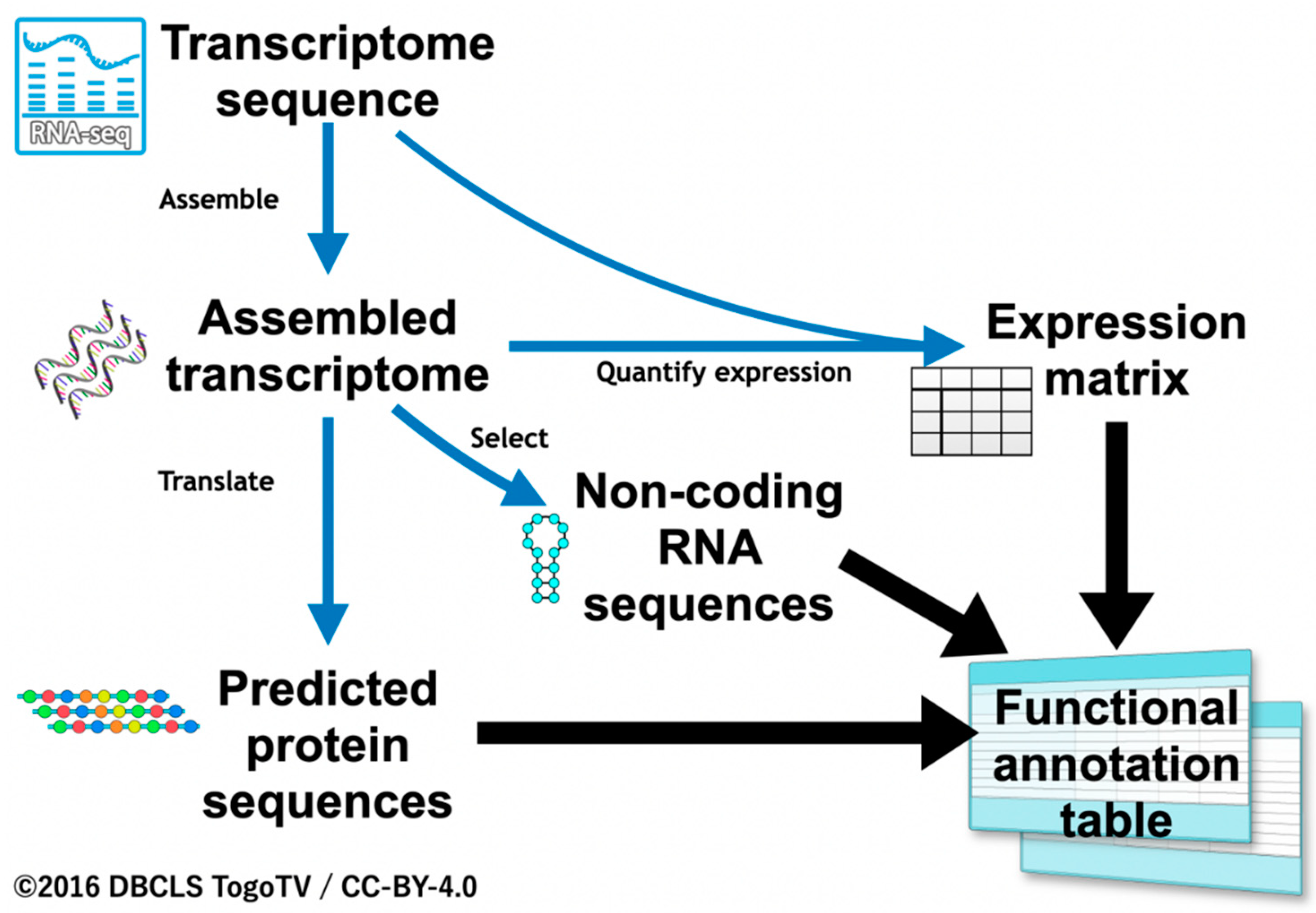
3. Results
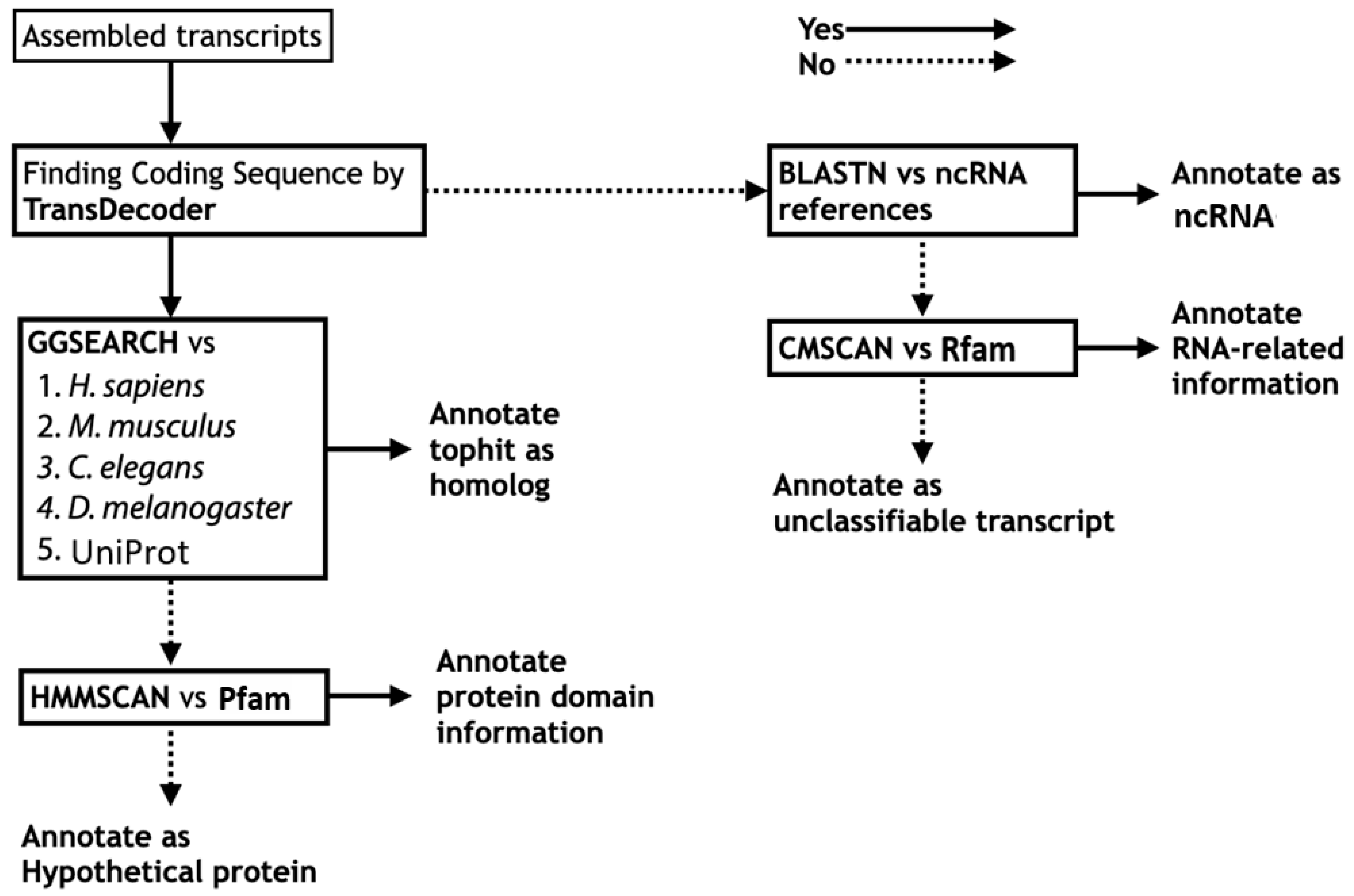
3.1. Functional Annotation Workflow for Insects
3.1.1. Functional Annotation of Coding Sequences
3.1.2. Functional Annotation of Non-Coding Sequences
3.1.3. Functional Annotation by Quantified Expression Information
3.2. Case Study for Fanflow4Insects
3.2.1. Functional Annotation of the Japanese Stick Insect Transcriptome Using Fanflow4Insects
3.2.2. Functional Annotation of Silkworm Using Fanflow4insects
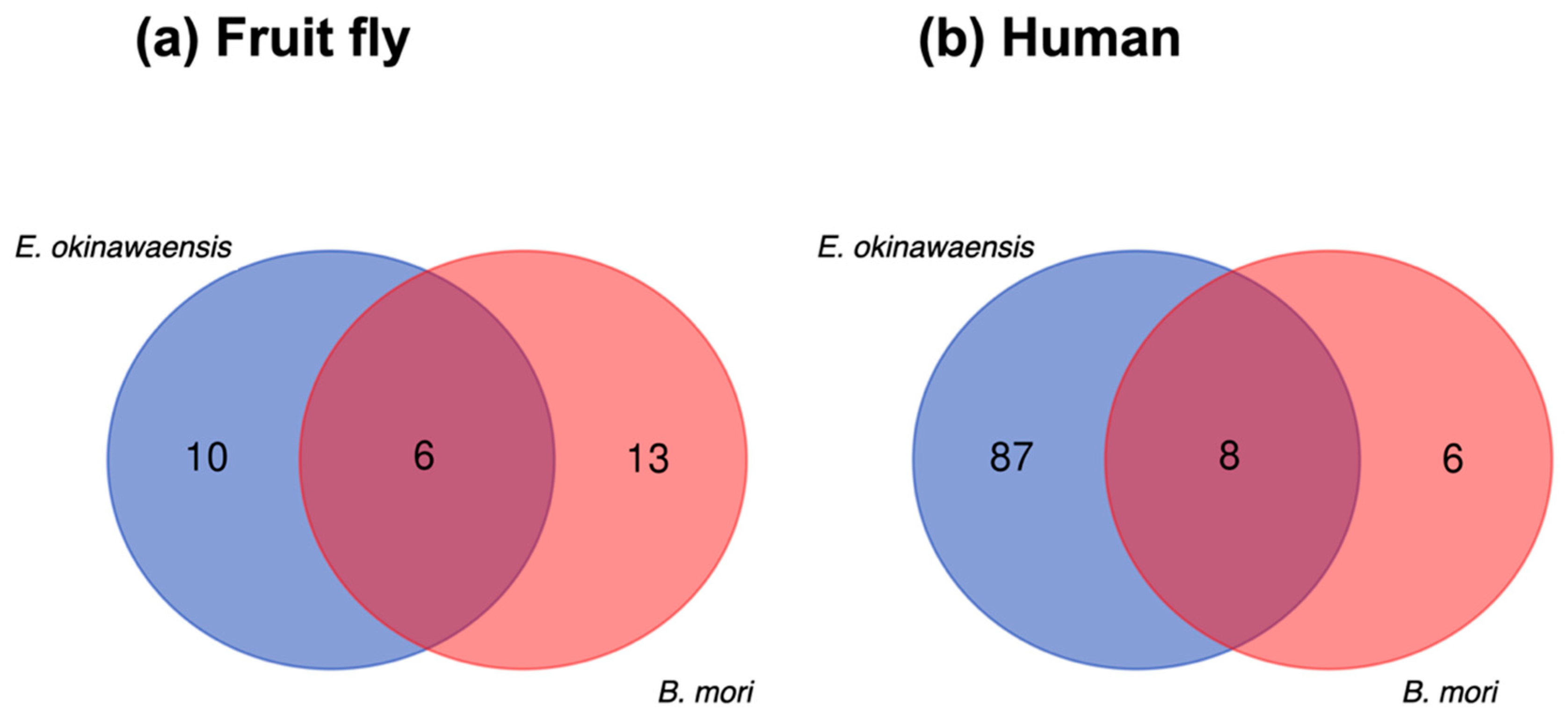
3.2.3. Comparison of ncRNAs between Japanese Stick Insect and Silkworm
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Genomes—NCBI Datasets for Insecta. Available online: https://www.ncbi.nlm.nih.gov/datasets/genomes/?taxon=50557 (accessed on 14 May 2022). [CrossRef]
- Hotaling, S.; Sproul, J.S.; Heckenhauer, J.; Powell, A.; Larracuente, A.M.; Pauls, S.U.; Kelley, J.L.; Frandsen, P.B. Long Reads Are Revolutionizing 20 Years of Insect Genome Sequencing. Genome Biol. Evol. 2021, 13, evab138. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, S.J.; Baker, R.H.; Simon, S.; DeSalle, R. We Can’t All Be Supermodels: The Value of Comparative Transcriptomics to the Study of Non-Model Insects. Insect Mol. Biol. 2015, 24, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Ewald, J.; Galvez, J.H.; Head, J.; Crump, D.; Bourque, G.; Basu, N.; Xia, J. Ultrafast Functional Profiling of RNA-Seq Data for Nonmodel Organisms. Genome Res. 2021, 31, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Larkin, A.; Marygold, S.J.; Antonazzo, G.; Attrill, H.; Dos Santos, G.; Garapati, P.V.; Goodman, J.L.; Gramates, L.S.; Millburn, G.; Strelets, V.B.; et al. FlyBase: Updates to the Drosophila Melanogaster Knowledge Base. Nucleic Acids Res. 2021, 49, D899–D907. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Yates, A.D.; Allen, J.; Amode, R.M.; Azov, A.G.; Barba, M.; Becerra, A.; Bhai, J.; Campbell, L.I.; Carbajo Martinez, M.; Chakiachvili, M.; et al. Ensembl Genomes 2022: An Expanding Genome Resource for Non-Vertebrates. Nucleic Acids Res. 2022, 50, D996–D1003. [Google Scholar] [CrossRef]
- Tabunoki, H.; Ono, H.; Ode, H.; Ishikawa, K.; Kawana, N.; Banno, Y.; Shimada, T.; Nakamura, Y.; Yamamoto, K.; Satoh, J.-I.; et al. Identification of Key Uric Acid Synthesis Pathway in a Unique Mutant Silkworm Bombyx Mori Model of Parkinson’s Disease. PLoS ONE 2013, 8, e69130. [Google Scholar] [CrossRef]
- Tabunoki, H.; Bono, H.; Ito, K.; Yokoyama, T. Can the Silkworm (Bombyx Mori) Be Used as a Human Disease Model? Drug Discov. Ther. 2016, 10, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, K.; Tsubota, T.; Jouraku, A.; Sezutsu, H.; Bono, H. Reference Transcriptome Data in Silkworm Bombyx Mori. Insects 2021, 12, 519. [Google Scholar] [CrossRef]
- Nojima, Y.; Bono, H.; Yokoyama, T.; Iwabuchi, K.; Sato, R.; Arai, K.; Tabunoki, H. Superoxide Dismutase Down-Regulation and the Oxidative Stress Is Required to Initiate Pupation in Bombyx Mori. Sci. Rep. 2019, 9, 14693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasukawa, T.; Furuno, M.; Nikaido, I.; Bono, H.; Hume, D.A.; Bult, C.; Hill, D.P.; Baldarelli, R.; Gough, J.; Kanapin, A.; et al. Development and Evaluation of an Automated Annotation Pipeline and CDNA Annotation System. Genome Res. 2003, 13, 1542–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, J.; Shinagawa, A.; Shibata, K.; Yoshino, M.; Itoh, M.; Ishii, Y.; Arakawa, T.; Hara, A.; Fukunishi, Y.; Konno, H.; et al. Functional Annotation of a Full-Length Mouse CDNA Collection. Nature 2001, 409, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, Y.; Furuno, M.; Kasukawa, T.; Adachi, J.; Bono, H.; Kondo, S. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 2002, 420, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The Transcriptional Landscape of the Mammalian Genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [Green Version]
- Maeda, N.; Kasukawa, T.; Oyama, R.; Gough, J.; Frith, M.; Engström, P.G.; Lenhard, B.; Aturaliya, R.N.; Batalov, S.; Beisel, K.W.; et al. Transcript Annotation in FANTOM3: Mouse Gene Catalog Based on Physical CDNAs. PLoS Genet. 2006, 2, e62. [Google Scholar] [CrossRef] [Green Version]
- Blaxter, M.; Danchin, A.; Savakis, B.; Fukami-Kobayashi, K.; Kurokawa, K.; Sugano, S.; Roberts, R.J.; Salzberg, S.L.; Wu, C.-I. Reminder to Deposit DNA Sequences. Science 2016, 352, 780. [Google Scholar] [CrossRef]
- Salzberg, S.L. Databases: Reminder to Deposit DNA Sequences. Nature 2016, 533, 179. [Google Scholar] [CrossRef] [Green Version]
- Bono, H. Meta-analysis of oxidative transcriptomes in insects. Antioxidants 2021, 10, 345. [Google Scholar] [CrossRef]
- Github Repository: Bonohu/SAQE. Available online: https://github.com/bonohu/SAQE (accessed on 14 May 2022).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- FASTA Sequence Comparison at the U. of Virginia. Available online: https://fasta.bioch.virginia.edu/ (accessed on 14 May 2022).
- HMMER: Biosequence Analysis Using Profile Hidden Markov Models. Available online: https://hmmer.org/ (accessed on 14 May 2022).
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-Fold Faster RNA Homology Searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded Coverage of Metagenomic, Viral and MicroRNA Families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, T.; Sasaki, S.; Yamaguchi, N.; Nakano, M.; Sato, H.; Iwabuchi, K.; Tabunoki, H.; Simpson, R.J.; Bono, H. De Novo Transcriptome Analysis for Examination of the Nutrition Metabolic System Related to the Evolutionary Process through Which Stick Insects Gain the Ability of Flight (Phasmatodea). BMC Res. Notes 2021, 14, 182. [Google Scholar] [CrossRef]
- Github Repository: FelixKrueger/TrimGalore. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 14 May 2022).
- UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Silkworm, Bombyx mori, Reference Transcriptome Data. Available online: https://dbarchive.biosciencedbc.jp/en/kaiko/data-2.html (accessed on 14 May 2022).
- Bono, H.; Ogata, H.; Goto, S.; Kanehisa, M. Reconstruction of Amino Acid Biosynthesis Pathways from the Complete Genome Sequence. Genome Res. 1998, 8, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, H.; Sakamoto, T.; Okochi, R.; Nishiko, M.; Sasaki, S.; Bono, H.; Tabunoki, H.; Iwabuchi, K. Apoptosis-mediated vasa down-regulation controls developmental transformation in Japanese Copidosoma floridanum female soldiers. Dev. Biol. 2019, 456, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Nishiko, M.; Bono, H.; Nakazato, T.; Yoshimura, J.; Tabunoki, H.; Iwabuchi, K. Analysis of molecular mechanism for acceleration of polyembryony using gene functional annotation pipeline in Copidosoma floridanum. BMC Genom. 2020, 21, 152. [Google Scholar] [CrossRef] [Green Version]
- Munro, S.A.; Lund, S.P.; Pine, P.S.; Binder, H.; Clevert, D.A.; Conesa, A.; Dopazo, J.; Fasold, M.; Hochreiter, S.; Hong, H.; et al. Assessing technical performance in differential gene expression experiments with external spike-in RNA control ratio mixtures. Nat. Comms. 2014, 5, 5125. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bioconda. Available online: https://bioconda.github.io (accessed on 14 May 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Name of Resource | URL |
|---|---|---|
| Protein sequences | Ensembl | 1 https://ftp.ensembl.org/pub/release-105/fasta/homo_sapiens/pep/Homo_sapiens.GRCh38.pep.all.fa.gz (accessed on 23 June 2022) |
| UniProtKB | https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz (accessed on 23 June 2022) | |
| Non-coding RNA sequences | Ensembl | 1 http://ftp.ensembl.org/pub/release-105/fasta/homo_sapiens/ncrna/Homo_sapiens.GRCh38.ncrna.fa.gz (accessed on 23 June 2022) |
| EnsemblGenomes | 1 http://ftp.ensemblgenomes.org/pub/release-52/metazoa/fasta/drosophila_melanogaster/ncrna/Drosophila_melanogaster.BDGP6.32.ncrna.fa.gz (accessed on 23 June 2022) | |
| Protein and RNA domain | Pfam | http://ftp.ebi.ac.uk/pub/databases/Pfam/releases/Pfam35.0/Pfam-A.hmm.gz (accessed on 23 June 2022) |
| Rfam | http://ftp.ebi.ac.uk/pub/databases/Rfam/14.7/Rfam.cm.gz (accessed on 23 June 2022) |
| Annotation Category | Annotation Level | Number of Annotations | Cumulative Number | Percentage |
|---|---|---|---|---|
| Protein homolog from tophit | Human or mouse homolog | 44,351 | 44,351 | 65.1 |
| C. elegans homolog | 3739 | 48,090 | 70.6 | |
| D. melanogaster homolog | 2349 | 50,439 | 74.0 | |
| Homolog found in UniProtKB | 4629 | 55,068 | 80.8 | |
| No protein homolog | Protein domain | 3808 | 58,876 | 86.4 |
| Hypothetical protein | 9286 | 68,162 | 100 |
| Annotation Level | All (311,357) | Hypothetical Protein (5699) | Unclassifiable Transcript (253,176) |
|---|---|---|---|
| Fat body-specific expression | 48,675 | 622 | 42,790 |
| Midgut-specific expression | 28,918 | 315 | 25,005 |
| Constitutive expression | 228,103 | 4606 | 181,521 |
| Not expressed | 5661 | 156 | 3860 |
| Annotation Category | Annotation Level | Number of Annotations | Cumulative Number | Percentage |
|---|---|---|---|---|
| Protein homolog from tophit | Human or mouse homolog | 31,354 | 31,354 | 68.6 |
| C. elegans homolog | 1752 | 33,106 | 72.4 | |
| D. melanogaster homolog | 2113 | 35,219 | 77.0 | |
| Homolog found in UniProtKB | 2888 | 38,107 | 83.4 | |
| No protein homolog | Protein domain | 5398 | 43,505 | 95.2 |
| Hypothetical protein | 2214 | 45,719 | 100 |
| Annotation Level | All (51,927) | Hypothetical Protein (1486) | Unclassifiable Transcript (12,845) |
|---|---|---|---|
| Fat body-specific expression | 39 | 1 | 11 |
| Midgut-specific expression | 108 | 0 | 29 |
| Malpighian tubule-specific expression | 83 | 3 | 14 |
| Silk gland-specific expression | 365 | 12 | 92 |
| Testis-specific expression | 861 | 19 | 492 |
| Ovary-specific expression | 179 | 9 | 55 |
| Constitutive expression | 7825 | 114 | 1205 |
| No expression | 609 | 24 | 125 |
| Transcript ID | Gene Name | Gene Description | |
|---|---|---|---|
| Annotation from fruit fly | FBtr0100888 | mt:lrRNA | mitochondrial large ribosomal RNA |
| FBtr0345722 | asRNA:CR45330 | antisense RNA:CR45330 | |
| FBtr0346876 | 28SrRNA: CR45837 | 28S ribosomal RNA:CR45837 | |
| FBtr0346877 | pre-rRNA:CR45846 | ribosomal RNA primary transcript:CR45846 | |
| FBtr0346881 | pre-rRNA:CR45847 | ribosomal RNA primary transcript:CR45847 | |
| FBtr0346882 | 18SrRNA: CR45841 | 18S ribosomal RNA:CR45841 | |
| Annotation from human | ENST00000450451 | novel transcript | |
| ENST00000501016 | novel transcript | ||
| ENST00000518947 | HOXA-AS3 | HOXA cluster antisense RNA 3 [Source:HGNC Symbol;Acc:HGNC:43748] | |
| ENST00000547387 | novel transcript, antisense to TUBA1B | ||
| ENST00000618978 | U2 | U2 spliceosomal RNA [Source:RFAM;Acc:RF00004] | |
| ENST00000623543 | novel transcript, antisense to TUBA8 | ||
| ENST00000631211 | novel transcript, similar to YY1 associated myogenesis RNA 1 YAM1 | ||
| ENST00000638356 | novel transcript, antisense to ATP4A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bono, H.; Sakamoto, T.; Kasukawa, T.; Tabunoki, H. Systematic Functional Annotation Workflow for Insects. Insects 2022, 13, 586. https://doi.org/10.3390/insects13070586
Bono H, Sakamoto T, Kasukawa T, Tabunoki H. Systematic Functional Annotation Workflow for Insects. Insects. 2022; 13(7):586. https://doi.org/10.3390/insects13070586
Chicago/Turabian StyleBono, Hidemasa, Takuma Sakamoto, Takeya Kasukawa, and Hiroko Tabunoki. 2022. "Systematic Functional Annotation Workflow for Insects" Insects 13, no. 7: 586. https://doi.org/10.3390/insects13070586