Tracing the Invasion and Expansion Characteristics of the Flatid Planthopper, Metcalfa pruinosa (Hemiptera: Flatidae), in Korea Using Mitochondrial DNA Sequences


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Primers, PCR, and Sequencing
2.3. Sequence Analysis
2.4. GenBank Data Search
2.5. Genetic Diversity
2.6. Structure
3. Results
3.1. DNA Barcoding Region
3.2. Worldwide COI Sequence Analysis
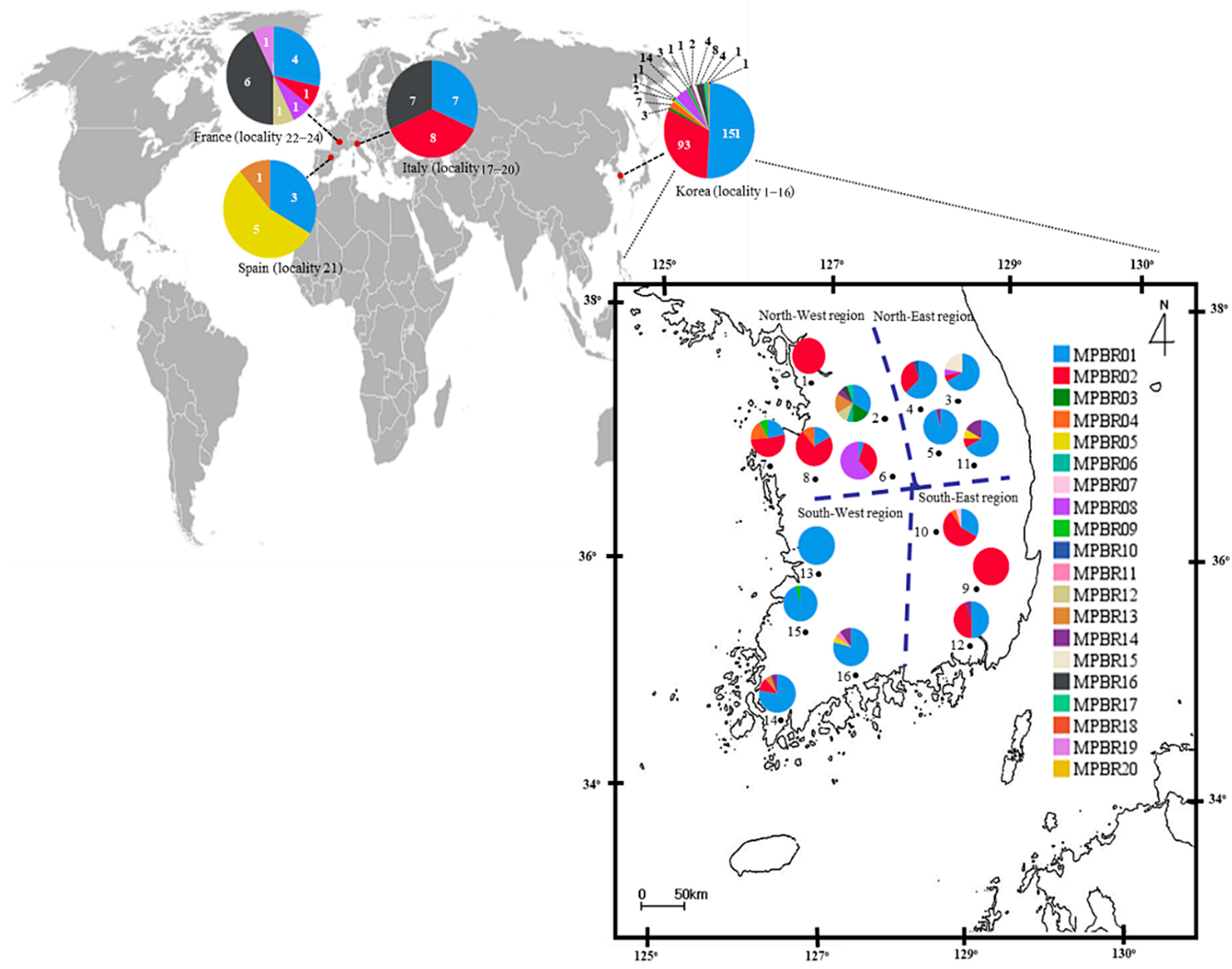
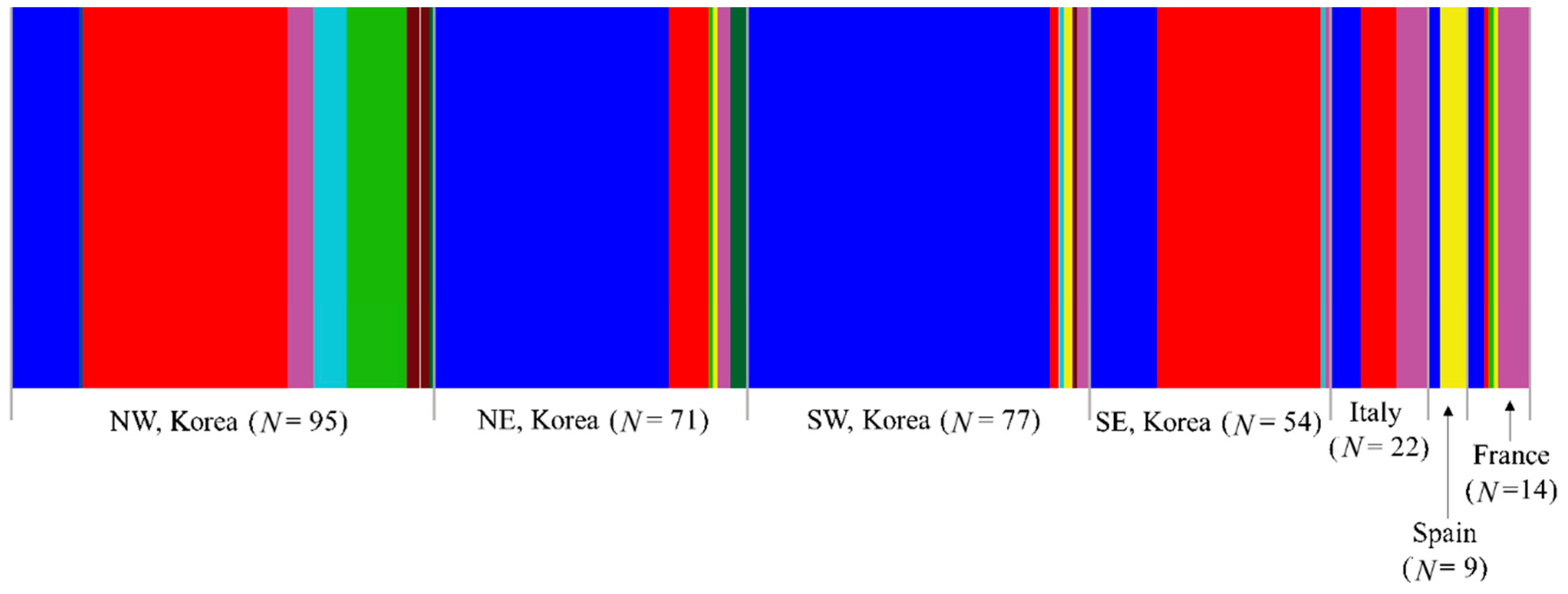
3.2.1. Haplotype Distribution
3.2.2. Genetic Diversity
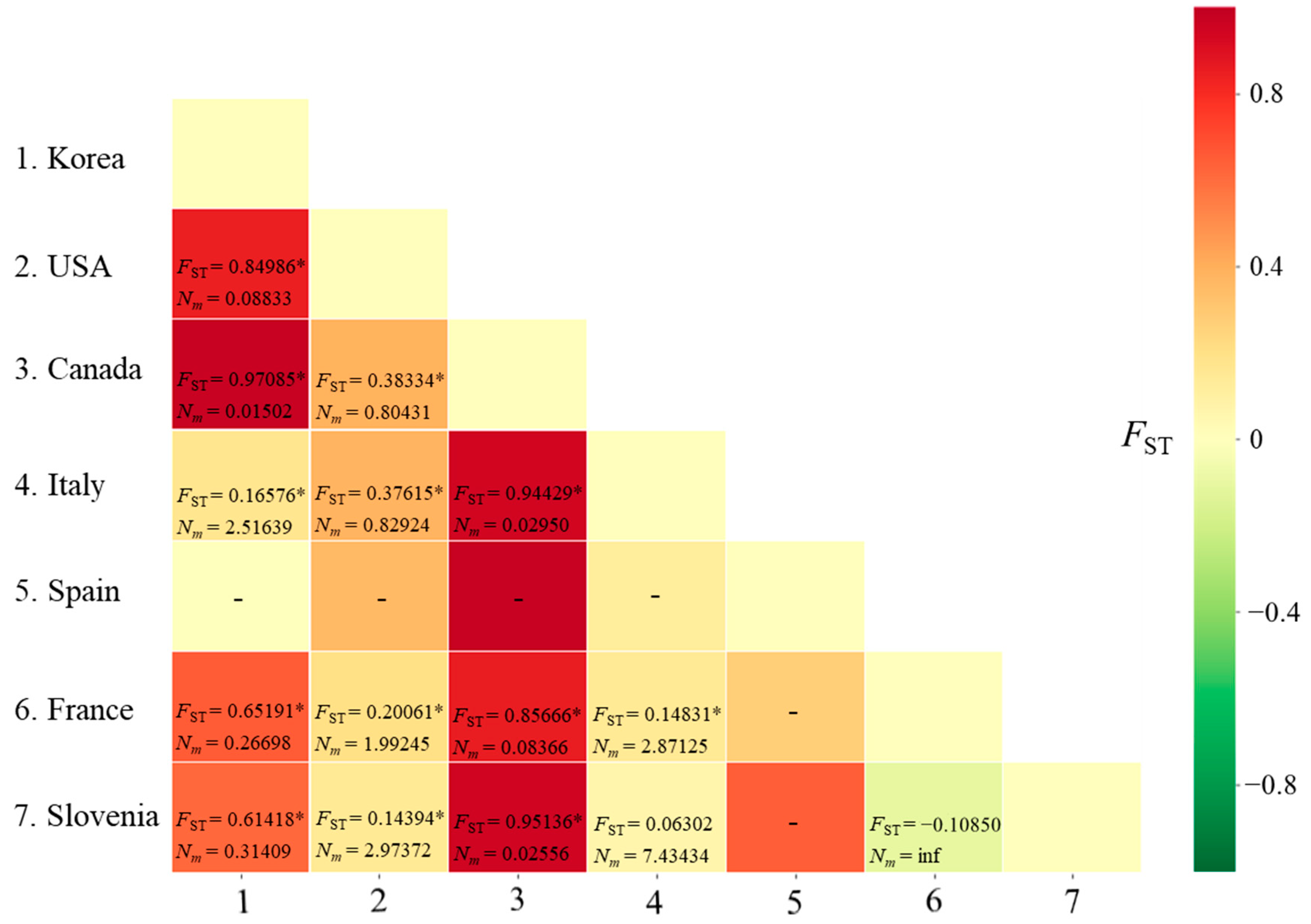
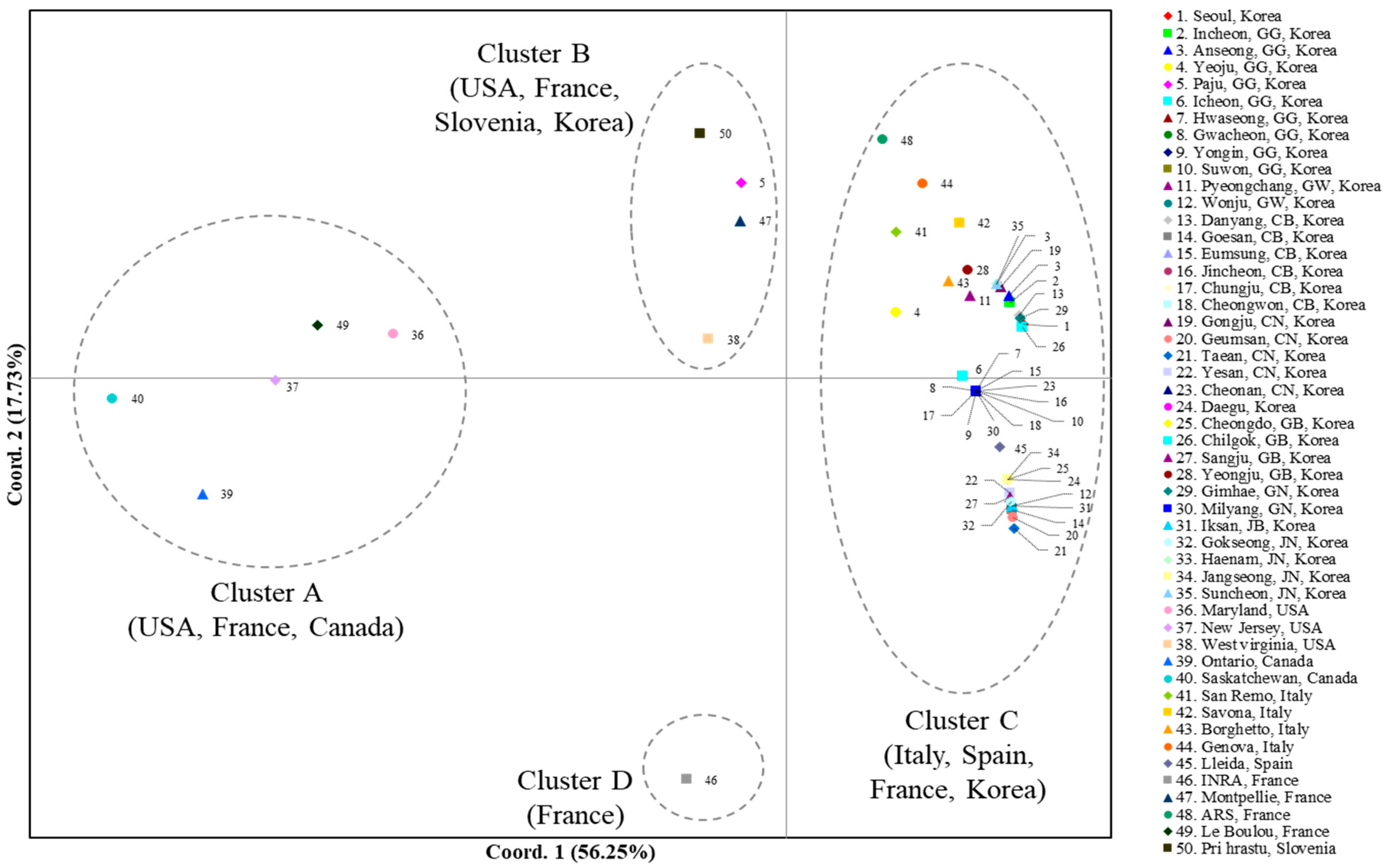
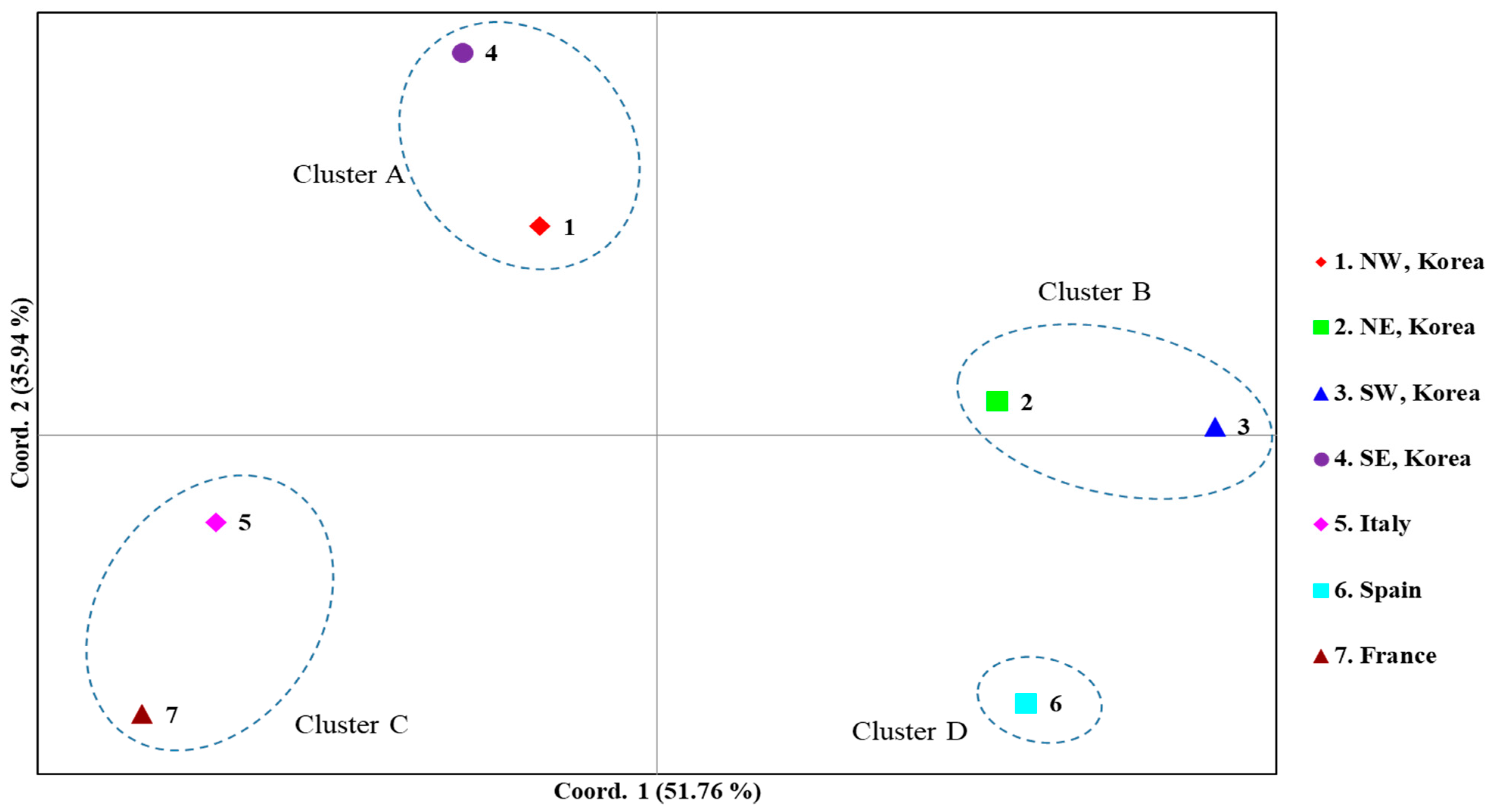
3.2.3. Genetic Distance and Structure
3.3. Variability in Region 5
3.4. Concatenated Sequences of the DNA Barcoding Region and Region 5
3.4.1. Sequence Analysis
3.4.2. Haplotype Distribution
3.4.3. Genetic Diversity
3.4.4. Genetic Distance
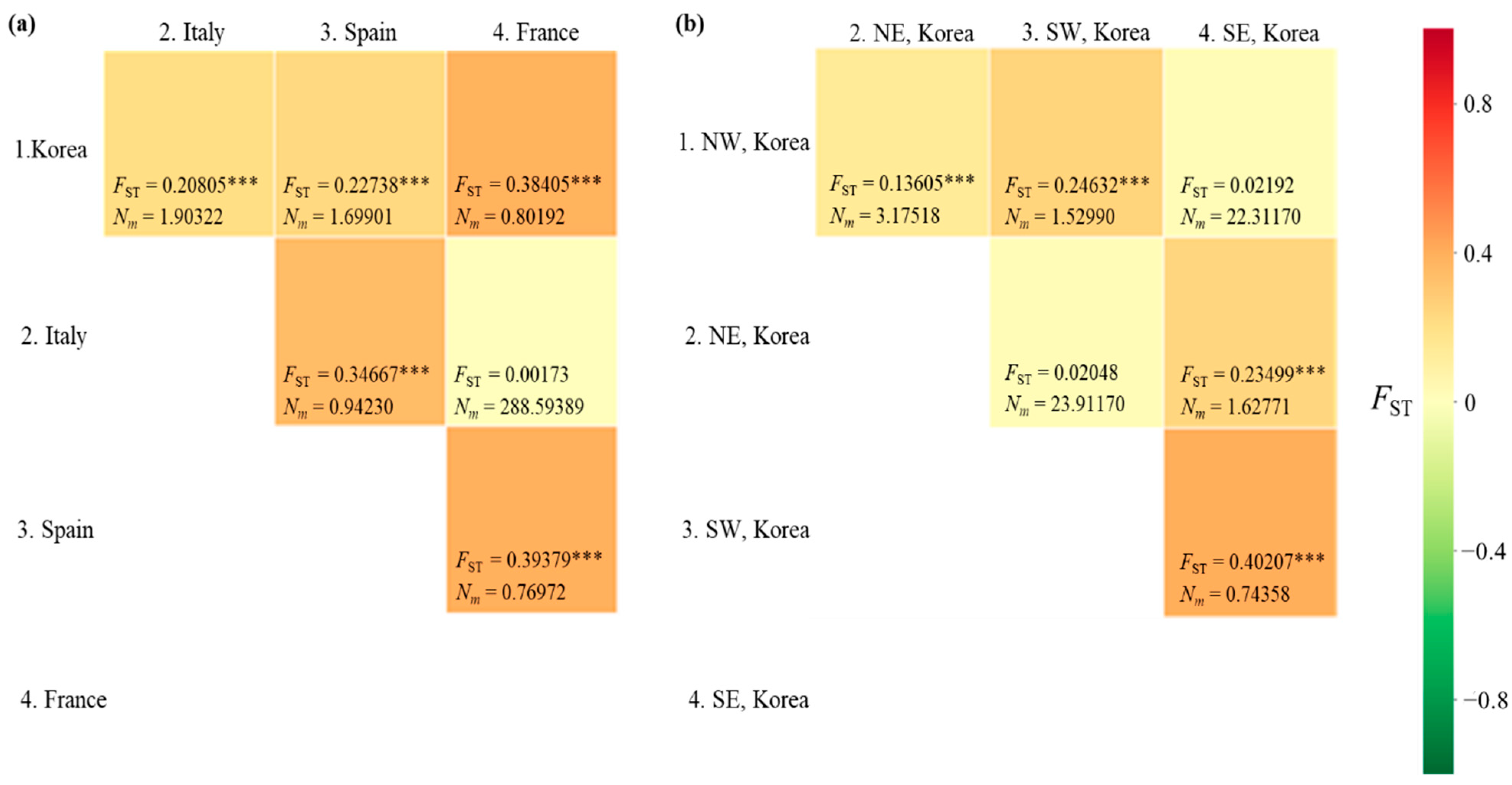
3.4.5. Structure
4. Discussion
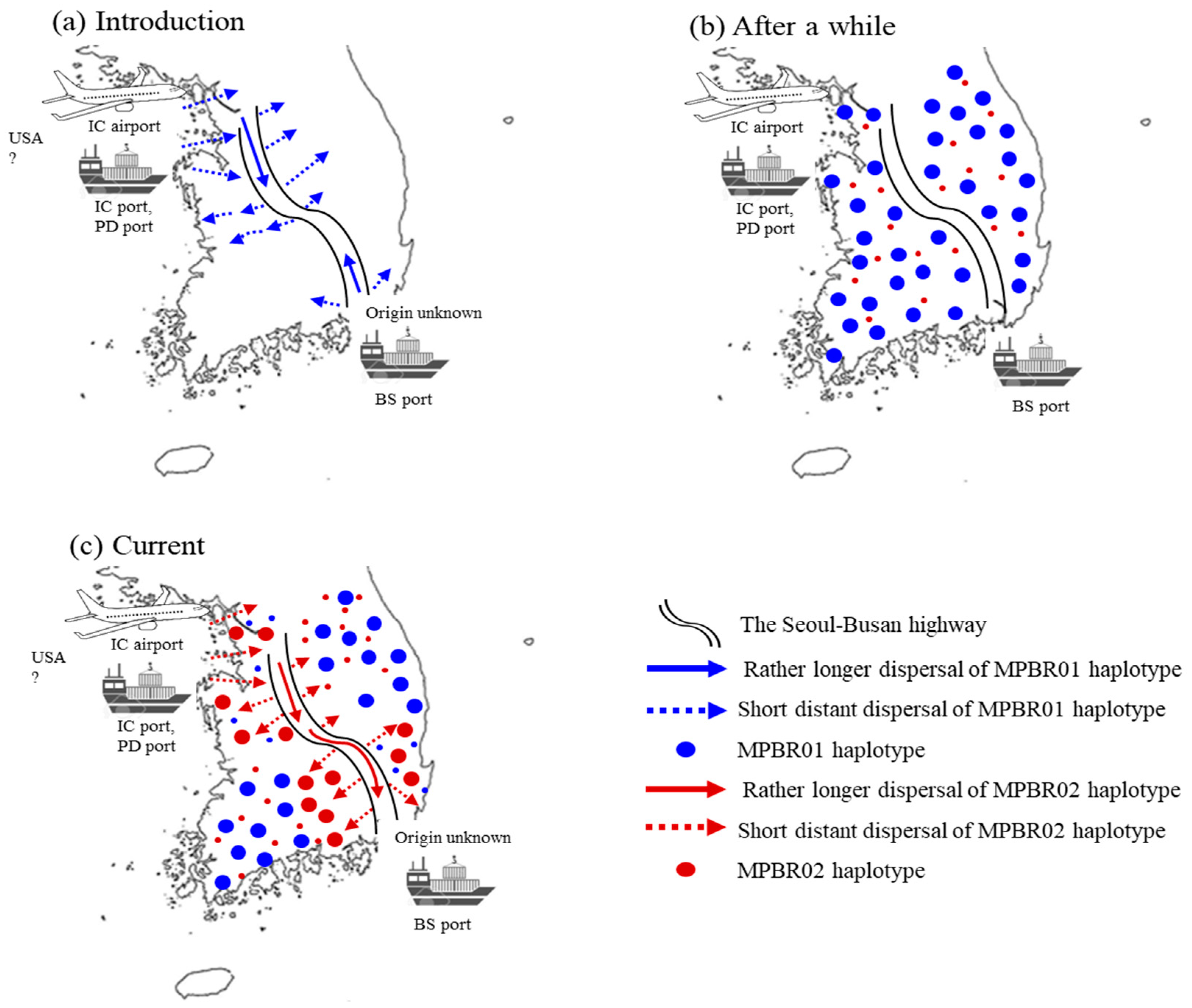
4.1. Origin and Point of Entry to Korea
4.2. Expansion Characteristics in Korea
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dean, H.A.; Bailey, J.C. A flatid planthopper, Metcalfa pruinosa. J. Econ. Entomol. 1961, 54, 1104–1106. [Google Scholar] [CrossRef]
- Wilson, S.W.; Lucchi, A. Distribution and ecology of Metcalfa pruinosa and associated planthoppers in North America (Homoptera: Fulgoroidea). In Atti dell’ Academia Nazionale Italiana di Entomologia, Rendiconti Anno; 2001; Volume 49, pp. 121–130. Available online: https://arpi.unipi.it/handle/11568/185729#.X-KsExapRPZ (accessed on 11 November 2020).
- Zangheri, S.; Donadini, P. Comparsa nel veneto di un omottero neartico: Metcalfa pruinosa Say (Homoptera, Flatidae). Redia 1980, 63, 301–305. [Google Scholar]
- EPPO. PQR Database; European and Mediterranean Plant Protection Organization: Paris, France, 2017; Available online: http://www.eppo.int/DATABASES/pqr/pqr.htmREF (accessed on 19 December 2020).
- Lee, H.S.; Wilson, S.W. First report of the Nearctic flatid planthopper Metcalfa pruinosa (Say) in the Republic of Korea (Hemiptera: Fulgoroidea). Entomol. News 2010, 121, 506–513. [Google Scholar] [CrossRef]
- Wilson, S.W.; McPherson, J.E. Life Histories of Anormenis septentrionalis, Metcalfa pruinosa, and Ormenoides venusta1 with descriptions of immature stages. Ann. Entomol. Soc. Am. 1981, 74, 299–311. [Google Scholar] [CrossRef]
- CABI. Crop Protection Compendium. Available online: http://www.cabicompendium.org/cpc/ (accessed on 4 April 2019).
- Kim, D.E.; Kil, J. Occurrence and host plant of Metcalfa pruinosa (Say) (Hemiptera: Flatidae) in Korea. J. Environ. Sci. Int. 2014, 23, 1385–1394. [Google Scholar] [CrossRef] [Green Version]
- Bagnoli, B.; Lucchi, A. Dannosit_a e misure di controllo integrato. In La Metcalfa Negli Ecostemi Italiani; Lucchi, A., Ed.; ARSIA Regione Toscana: Firenze, Italy, 2000; pp. 65–88. [Google Scholar]
- Wilson, S.W.; Lucchi, A. Aspetti Sistematici, Corologici, Ecologici. In La Metcalfa Negli Ecosistemi Italiani; Lucchi, A., Ed.; Agenzia Regionale per lo Svilluppo e l’Innovazione nel settore Agricolo-forestale: Firenze, Italy, 2000; p. 163. [Google Scholar]
- Wilson, S.W.; Lucchi, A. Feeding activity of the flatid planthopper Metcalfa pruinosa (Homoptera: Fulgoroidea). J. Kans. Entomol. Soc. 2007, 80, 175–178. [Google Scholar] [CrossRef]
- Lauterer, P. A New Pest of Ornamental Horticulture in the Czech Republic. Plant Prot. Sci. 2002, 38, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Alma, A.; Ferracini, C.; Burgio, G. Development of a sequential plan to evaluate Neodryinus typhlocybae (Ashmead) (Hymenoptera: Dryinidae) population associated with Metcalfa pruinosa (Say) (Homoptera: Flatidae) infestation in northwestern Italy. Environ. Entomol. 2005, 34, 819–824. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kim, M.; Hong, K.J.; Lee, S. Outbreak of an exotic flatid, Metcalfa pruinosa (Say) (Hemiptera: Flatidae), in the capital region of Korea. J. Asia Pac. Entomol. 2011, 14, 473–478. [Google Scholar] [CrossRef]
- Ciampolini, M.; Grossi, A.; Zottarelli, G. Damage to soyabean through attack by Metcalfa pruinosa. L’Inf. Agrar. 1987, 43, 101–103. [Google Scholar]
- Kwon, D.H.; Kim, M.; Kim, H.; Lee, Y.; Hong, K.J.; Lee, S.H.; Lee, S. Estimation of genetic divergence based on mitochondrial DNA variation for an invasive alien species, Metcalfa pruinosa (Say), in Korea. J. Asia Pac. Entomol. 2015, 18, 447–451. [Google Scholar] [CrossRef]
- Park, C.G.; Min, S.; Lee, G.S.; Kim, S.; Lee, Y.; Lee, S.; Hong, K.J.; Wilson, S.W.; Akimoto, S.I.; Lee, W. Genetic variability of the invasive species Metcalfa pruinosa (Hemiptera: Flatidae) in the Republic of Korea. J. Econ. Entomol. 2016, 109, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Gwiazdowski, R.A.; Foottit, R.G.; Maw, H.E.; Hebert, P.D. The Hemiptera (Insecta) of Canada: Constructing a reference library of DNA barcodes. PLoS ONE 2015, 10, e0125635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, A.R.; Kim, K.G.; Park, J.W.; Kim, I. Genetic diversity of the dung beetle, Copris tripartitus (Coleoptera: Scarabaeidae), that is endangered in Korea. Entomol. Res. 2012, 42, 247–261. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, G.H.; Park, J.S.; Jeong, J.S.; Jeong, N.R.; Lee, W.; Kim, I. Genomic comparison and phylogenetic inference in Fulgoroidea with the complete mitochondrial genome sequences of the flatid planthoppers, Metcalfa pruinosa and Salurnis marginella (Hemiptera: Flatidae). Genomics 2020. under review. [Google Scholar]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP* Phylogenetic Analysis Using Parsimony (*and Other Method) Version 4.10; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Hebert, P.D.N.; Ratnasingham, S.; Zakharov, E.V.; Levesque-Beaudin, V.; Telfer, A.C.; Milton, M.A. Counting animal species with DNA barcodes: Canadian insects. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M. Genetic variation within species. In Molecular Evolutionary Genetics; Nei, M., Ed.; Columbia University Press: New York, NY, USA, 1987; Chapter 8; pp. 176–207. [Google Scholar]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Orlóci, L. Multivariate Analysis in Vegetation Research; Springer: Dordrecht, The Netherlands, 1978. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corander, J.; Tang, J. Bayesian analysis of population structure based on linked molecular information. Math. Biosci. 2007, 205, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.S.; Park, J.S.; Kim, M.J.; Kim, J.S.; Jeong, S.Y.; Jeong, J.S.; Park, J.; Kim, I. Geographic variation in the spotted-wing drosophila, Drosophila suzukii (Diptera: Drosophilidae), based on mitochondrial DNA sequences. Mitochondrial DNA Part A 2018, 29, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Watterson, G.A.; Guess, H.A. Is the most frequent allele the oldest? Theor. Popul. Biol. 1977, 11, 141–160. [Google Scholar] [CrossRef]
- Sakai, A.K.; Allendorf, F.W.; Holt, J.S.; Lodge, D.M.; Molofsky, J.; With, K.A.; Baughman, S.; Cabin, R.J.; Cohen, J.E.; Ellstrand, N.C.; et al. The population biology of invasive species. Annu. Rev. Ecol. Syst. 2001, 32, 305–332. [Google Scholar] [CrossRef] [Green Version]
- Dlugosch, K.M.; Parker, I.M. Founding events in species invasions: Genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 2008, 17, 431–449. [Google Scholar] [CrossRef]
- Puillandre, N.; Dupas, S.; Dangles, O.; Zeddam, J.L.; Capdevielle-Dulac, C.; Barbin, K.; Torres-Leguizamon, M.; Silvain, J.F. Genetic bottleneck in invasive species: The potato tuber moth adds to the list. Biol. Invasions 2008, 10, 319–333. [Google Scholar] [CrossRef]
- Pantaleoni, R.A. Modalita d’invasione di nuovo areale in Metcalfa pruinosa (Say) (Au che—norrhynca Flatidae). In Bollettino Dell’istituto di Entomologia “Guido Grandi” Della Università Degli Studi di Bolognoa; Tip. Compositori: Bologna, Italy, 1989; Volume 43, pp. 1–7. [Google Scholar]
- Suarez, A.V.; Holway, D.A.; Case, T.J. Patterns of spread in biological invasions dominated by long-distance jump dispersal: Insights from Argentine ants. Proc. Natl. Acad. Sci. USA 2001, 98, 1095–1100. [Google Scholar] [CrossRef] [Green Version]
- Muirhead, J.R.; Leung, B.; van Overdijk, C.; Kelly, D.W.; Nandakumar, K.; Marchant, K.R.; MacIsaac, H.J. Modelling local and long-distance dispersal of invasive emerald ash borer Agrilus planipennis (Coleoptera) in North America. Divers. Distrib. 2006, 12, 71–79. [Google Scholar] [CrossRef]
- Wilson, J.R.; Dormontt, E.E.; Prentis, P.J.; Lowe, A.J.; Richardson, D.M. Something in the way you move: Dispersal pathways affect invasion success. Trends Ecol. Evol. 2009, 24, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Bullock, J.M.; Bonte, D.; Pufal, G.; da Silva Carvalho, C.; Chapman, D.S.; García, C.; García, D.; Matthysen, E.; Delgado, M.M. Human-mediated dispersal and the rewiring of spatial networks. Trends Ecol. Evol. 2018, 33, 958–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, I.; Pinto, M.A.; De la Rúa, P. Temporal changes in mitochondrial diversity highlights contrasting population events in Macaronesian honey bees. Apidologie 2013, 44, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Legg, J.P.; Sseruwagi, P.; Boniface, S.; Okao-Okuja, G.; Shirima, R.; Bigirimana, S.; Gashaka, G.; Herrmann, H.W.; Jeremiah, S.; Obiero, H.; et al. Spatio-temporal patterns of genetic change amongst populations of cassava Bemisia tabaci whiteflies driving virus pandemics in East and Central Africa. Virus Res. 2014, 186, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Moule, H.; Chaplin, K.; Bray, R.D.; Miller, K.A.; Thompson, M.B.; Chapple, D.G. A matter of time: Temporal variation in the introduction history and population genetic structuring of an invasive lizard. Curr. Zool. 2015, 61, 456–464. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Haplotype | Locality | |||||||
|---|---|---|---|---|---|---|---|---|
| 1. Korea (659) | 2. USA (36) | 3. Canada (64) | 4. Italy (32) | 5. Spain (14) | 6. France (21) | 7. Slovenia (4) | Total | |
| 1. MPH01 | 0.943 (622) | 0.028 (1) | 0.781 (25) | 1.000 (14) | 0.476 (10) | 0.500 (2) | 0.812 (674) | |
| 2. MPH02 | 0.050 (33) | 0.111 (4) | 0.219 (7) | 0.381 (8) | 0.500 (2) | 0.065 (54) | ||
| 3. MPH03 | 0.006 (4) | 0.028 (1) | 0.006 (5) | |||||
| 4 MPH04 | 0.028 (1) | 0.001 (1) | ||||||
| 5. MPH05 | 0.056 (2) | 0.002 (2) | ||||||
| 6. MPH06 | 0.028 (1) | 0.001 (1) | ||||||
| 7. MPH07 | 0.056 (2) | 0.002 (2) | ||||||
| 8. MPH08 | 0.111 (4) | 0.005 (4) | ||||||
| 9. MPH09 | 0.028 (1) | 0.001 (1) | ||||||
| 10. MPH10 | 0.111 (4) | 0.938 (60) | 0.077 (64) | |||||
| 11. MPH11 | 0.028 (1) | 0.001 (1) | ||||||
| 12. MPH12 | 0.143 (3) | 0.004 (3) | ||||||
| 13. MPH13 | 0.028 (1) | 0.001 (1) | ||||||
| 14. MPH14 | 0.056 (2) | 0.002 (2) | ||||||
| 15. MPH15 | 0.139 (5) | 0.006 (5) | ||||||
| 16. MPH16 | 0.083 (3) | 0.004 (3) | ||||||
| 17. MPH17 | 0.083 (3) | 0.004 (3) | ||||||
| 18. MPH18 | 0.016 (1) | 0.001 (1) | ||||||
| 19. MPH19 | 0.031 (2) | 0.002 (2) | ||||||
| 20. MPH20 | 0.016 (1) | 0.001 (1) | ||||||
| Total | 0.794 (659) | 0.043 (36) | 0.077 (64) | 0.039 (32) | 0.019 (14) | 0.025 (21) | 0.005 (4) | |
| Locality | SS a | NH b | Hc | NP d | MSD e (%) | MPD f | π g |
|---|---|---|---|---|---|---|---|
| 1. Korea | 297 | 17 | 0.6414 ± 0.0209 | 13 | 0.92 | 1.958367 | 0.001803 ± 0.001133 |
| NW, Korea | 95 | 12 | 0.7118 ± 0.0416 | 11 | 0.92 | 2.407615 | 0.002217 ± 0.001346 |
| NE, Korea | 71 | 7 | 0.4306 ± 0.0700 | 11 | 0.92 | 1.814085 | 0.001670 ± 0.001080 |
| SW, Korea | 77 | 7 | 0.2201 ± 0.0629 | 11 | 0.92 | 1.013671 | 0.000933 ± 0.000702 |
| SE, Korea | 54 | 5 | 0.4864 ± 0.0584 | 10 | 0.83 | 1.217331 | 0.001121 ± 0.000804 |
| 2. Italy | 22 | 3 | 0.6970 ± 0.0324 | 9 | 0.83 | 4.090909 | 0.003767 ± 0.002177 |
| 3. Spain | 9 | 3 | 0.6389 ± 0.1258 | 3 | 0.28 | 1.000000 | 0.000924 ± 0.000775 |
| 4. France | 14 | 6 | 0.7692 ± 0.0895 | 13 | 0.92 | 5.395604 | 0.004968 ± 0.002860 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.H.; Jeong, J.S.; Park, J.S.; Kim, M.J.; Jeong, N.R.; Jeong, S.Y.; Lee, G.S.; Lee, W.; Kim, I. Tracing the Invasion and Expansion Characteristics of the Flatid Planthopper, Metcalfa pruinosa (Hemiptera: Flatidae), in Korea Using Mitochondrial DNA Sequences. Insects 2021, 12, 4. https://doi.org/10.3390/insects12010004
Lee KH, Jeong JS, Park JS, Kim MJ, Jeong NR, Jeong SY, Lee GS, Lee W, Kim I. Tracing the Invasion and Expansion Characteristics of the Flatid Planthopper, Metcalfa pruinosa (Hemiptera: Flatidae), in Korea Using Mitochondrial DNA Sequences. Insects. 2021; 12(1):4. https://doi.org/10.3390/insects12010004
Chicago/Turabian StyleLee, Keon Hee, Jun Seong Jeong, Jeong Sun Park, Min Jee Kim, Na Ra Jeong, Su Yeon Jeong, Gwan Seok Lee, Wonhoon Lee, and Iksoo Kim. 2021. "Tracing the Invasion and Expansion Characteristics of the Flatid Planthopper, Metcalfa pruinosa (Hemiptera: Flatidae), in Korea Using Mitochondrial DNA Sequences" Insects 12, no. 1: 4. https://doi.org/10.3390/insects12010004