Applications of CRISPR/Cas9 for the Treatment of Duchenne Muscular Dystrophy



Abstract
:1. Introduction
2. Theory of CRISPR Application for Gene Editing
3. DMD Studies In Vitro and Alternatives to Viral Delivery of CRISPR
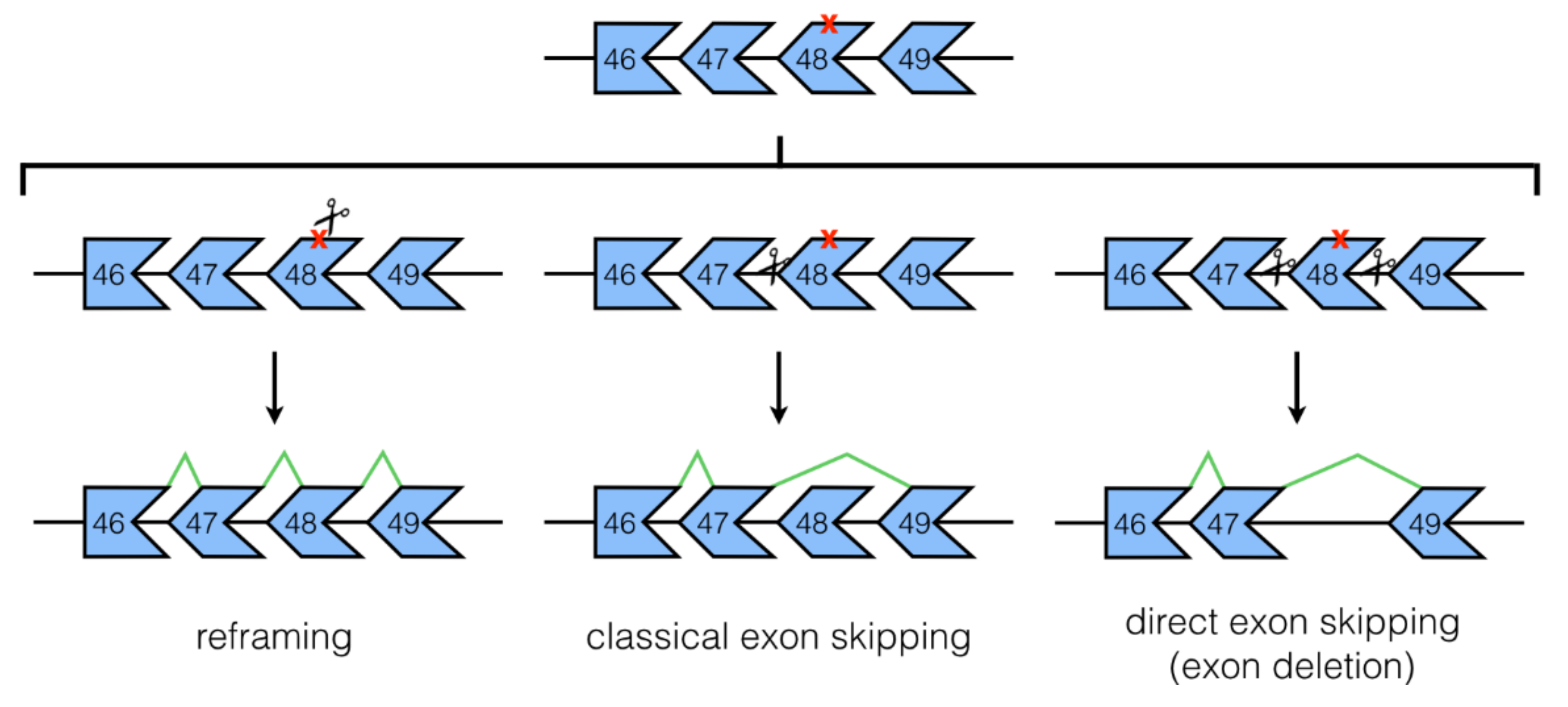
3.1. NHEJ Mechanisms: Classical and Direct Exon Skipping
3.2. Base Editing
3.3. Utrophin Upregulation
4. DMD Studies In Vivo
4.1. Studies in DMD Mouse Models
4.2. Study in a DMD Dog Model
4.3. CRISPR/Cas9 for the Generation of DMD Animal/Cell Models
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- O’Brien, K.F.; Kunkel, L.M. Dystrophin and muscular dystrophy: Past, present, and future. Mol. Genet. Metab. 2001, 74, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P.; Kahl, S.D. Association of dystrophin and an integral membrane glycoprotein. Nature 1989, 338, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Manzur, A.Y.; Kinali, M.; Muntoni, F. Update on the management of duchenne muscular dystrophy. Arch. Dis. Child. 2008, 93, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Landfeldt, E.; Sejersen, T.; Tulinius, M. A mini review and implementation model for using ataluren to treat nonsense mutation duchenne muscular dystrophy. Acta Paediatr. 2018. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Tyagi, R.; Mohanty, M.; Goyal, M.; Silva, K.R.; Wijekoon, N. Dystrophin induced cognitive impairment: Mechanisms, models and therapeutic strategies. Ann. Neurosci. 2015, 22, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Mah, J.K. Current and emerging treatment strategies for duchenne muscular dystrophy. Neuropsychiatr. Dis. Treat. 2016, 12, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.G.; Coffey, A.J.; Bobrow, M.; Bentley, D.R. Exon structure of the human dystrophin gene. Genomics 1993, 16, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Ervasti, J.M. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta 2007, 1772, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Abbs, S.; Tuffery-Giraud, S.; Bakker, E.; Ferlini, A.; Sejersen, T.; Mueller, C.R. Best practice guidelines on molecular diagnostics in duchenne/becker muscular dystrophies. Neuromuscul. Disord. 2010, 20, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Aslesh, T.; Maruyama, R.; Yokota, T. Skipping multiple exons to treat DMD-promises and challenges. Biomedicines 2018, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Yokota, T. Invention and early history of exon skipping and splice modulation. Methods Mol. Biol. 2018, 1828, 3–30. [Google Scholar] [PubMed]
- Lehto, T.; Castillo Alvarez, A.; Gauck, S.; Gait, M.J.; Coursindel, T.; Wood, M.J.; Lebleu, B.; Boisguerin, P. Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells. Nucleic Acids Res. 2014, 42, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [PubMed]
- O’Connell, M.R.; Oakes, B.L.; Sternberg, S.H.; East-Seletsky, A.; Kaplan, M.; Doudna, J.A. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 2014, 516, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Nakade, S.; Yamamoto, T.; Sakuma, T. Cas9, Cpf1 and C2C1/2/3-What’s next? Bioengineered 2017, 8, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedland, A.E.; Baral, R.; Singhal, P.; Loveluck, K.; Shen, S.; Sanchez, M.; Marco, E.; Gotta, G.M.; Maeder, M.L.; Kennedy, E.M.; et al. Characterization of staphylococcus aureus Cas9: A smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015, 16, 257. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Tang, L.; He, X.; Liu, X.; Zhou, C.; Liu, J.; Ge, X.; Li, J.; Liu, C.; Zhao, J.; et al. SaCas9 requires 5′-NNGRRT-3′ PAM for sufficient cleavage and possesses higher cleavage activity than SpCas9 or FnCpf1 in human cells. Biotechnol. J. 2018, 13, e1800080. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M. Studying cardiac arrhythmias in the mouse—A reasonable model for probing mechanisms? Trends Cardiovasc. Med. 2004, 14, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Salama, G.; London, B. Mouse models of long QT syndrome. J. Physiol. 2007, 578, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Denning, C.; Borgdorff, V.; Crutchley, J.; Firth, K.S.; George, V.; Kalra, S.; Kondrashov, A.; Hoang, M.D.; Mosqueira, D.; Patel, A.; et al. Cardiomyocytes from human pluripotent stem cells: From laboratory curiosity to industrial biomedical platform. Biochim. Biophys. Acta 2016, 1863, 1728–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, P.S.; Keenan, R.E.; Swartout, J.C. Characterizing interspecies uncertainty using data from studies of anti-neoplastic agents in animals and humans. Toxicol. Appl. Pharmacol. 2008, 233, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wei, G.; Li, P.; Zhou, X.; Zhang, Y. Urine-derived stem cells: A novel and versatile progenitor source for cell-based therapy and regenerative medicine. Genes Dis. 2014, 1, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The treat-nmd DMD global database: Analysis of more than 7000 duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by talen and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtal, D.; Kemaladewi, D.U.; Malam, Z.; Abdullah, S.; Wong, T.W.; Hyatt, E.; Baghestani, Z.; Pereira, S.; Stavropoulos, J.; Mouly, V.; et al. Spell checking nature: Versatility of CRISPR/Cas9 for developing treatments for inherited disorders. Am. J. Hum. Genet. 2016, 98, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Iyombe-Engembe, J.P.; Ouellet, D.L.; Barbeau, X.; Rousseau, J.; Chapdelaine, P.; Lague, P.; Tremblay, J.P. Efficient restoration of the dystrophin gene reading frame and protein structure in DMD myoblasts using the cindel method. Mol. Ther. Nucleic Acids 2016, 5, e283. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Stefanucci, L.; Janssen, J.M.; Liu, J.; Chen, X.; Mouly, V.; Goncalves, M.A. Selection-free gene repair after adenoviral vector transduction of designer nucleases: Rescue of dystrophin synthesis in DMD muscle cell populations. Nucleic Acids Res. 2016, 44, 1449–1470. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Park, K.H.; Zhao, L.; Xu, J.; El Refaey, M.; Gao, Y.; Zhu, H.; Ma, J.; Han, R. Crispr-mediated genome editing restores dystrophin expression and function in MDX mice. Mol. Ther. 2016, 24, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Liu, J.; Janssen, J.M.; Chen, X.; Goncalves, M.A. Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci. Rep. 2016, 6, 37051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Long, C.; Li, H.; McAnally, J.R.; Baskin, K.K.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017, 3, e1602814. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, A.; Duguez, S.; Moiani, A.; Izmiryan, A.; Barbon, E.; Martin, S.; Mamchaoui, K.; Mouly, V.; Bernardi, F.; Mavilio, F.; et al. Correction of the exon 2 duplication in DMD myoblasts by a single CRISPR/Cas9 system. Mol. Ther. Nucleic Acids 2017, 7, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wu, F.; Mosenson, J.; Zhang, H.; He, T.C.; Wu, W.S. Crispr/Cas9-mediated genome editing corrects dystrophin mutation in skeletal muscle stem cells in a mouse model of muscle dystrophy. Mol. Ther. Nucleic Acids 2017, 7, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Mokhonova, E.; Quinonez, M.; Pyle, A.D.; Spencer, M.J. Creation of a novel humanized dystrophic mouse model of duchenne muscular dystrophy and application of a CRISPR/Cas9 gene editing therapy. J. Neuromuscul. Dis. 2017, 4, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.H.; Bassel-Duby, R.; Olson, E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2017, 2, e95918. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; McAnally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 9, eaan8081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrke-Schulz, E.; Schiwon, M.; Leitner, T.; Dávid, S.; Bergmann, T.; Liu, J.; Ehrhardt, A. Crispr/Cas9 delivery with one single adenoviral vector devoid of all viral genes. Sci. Rep. 2017, 7, 17113. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Li, H.; Tiburcy, M.; Rodriguez-Caycedo, C.; Kyrychenko, V.; Zhou, H.; Zhang, Y.; Min, Y.L.; Shelton, J.M.; Mammen, P.P.A.; et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018, 4, eaap9004. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Lu-Nguyen, N.B.; Malerba, A.; Kim, E.; Kim, D.; Cappellari, O.; Cho, H.Y.; Dickson, G.; Popplewell, L.; Kim, J.S. Functional rescue of dystrophin deficiency in mice caused by frameshift mutations using campylobacter jejuni Cas9. Mol. Ther. 2018, 26, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Duchene, B.L.; Cherif, K.; Iyombe-Engembe, J.P.; Guyon, A.; Rousseau, J.; Ouellet, D.L.; Barbeau, X.; Lague, P.; Tremblay, J.P. CRISPR-induced deletion with saCas9 restores dystrophin expression in dystrophic models in vitro and in vivo. Mol. Ther. 2018, 26, 2604–2616. [Google Scholar] [CrossRef] [PubMed]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.-M.; Koo, T.; Kim, K.; Lim, K.; Baek, G.; Kim, S.-T.; Kim, H.S.; Kim, D.-E.; Lee, H.; Chung, E.; et al. Adenine base editing in mouse embryos and an adult mouse model of duchenne muscular dystrophy. Nat. Biotechnol. 2018, 36, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ma, Y.; Huang, T.; Chen, Y.; Peng, Y.; Li, B.; Li, J.; Zhang, Y.; Song, B.; Sun, X.; et al. Genetic modulation of RNA splicing with a CRISPR-guided cytidine deaminase. Mol. Cell 2018, 72, 380–394.e7. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yokota, T.; Wood, M.J. Development of multiexon skipping antisense oligonucleotide therapy for duchenne muscular dystrophy. Biomed. Res. Int. 2013, 2013, 402369. [Google Scholar] [CrossRef] [PubMed]
- Beroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizard, M.P.; Voelckel, M.A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with duchenne muscular dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Duddy, W.; Partridge, T. Optimizing exon skipping therapies for DMD. Acta Myol. 2007, 26, 179–184. [Google Scholar] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Khurana, T.S.; Watkins, S.C.; Chafey, P.; Chelly, J.; Tome, F.M.; Fardeau, M.; Kaplan, J.C.; Kunkel, L.M. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul. Disord. 1991, 1, 185–194. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Ervasti, J.M.; Matsumura, K.; Kahl, S.D.; Leveille, C.J.; Campbell, K.P. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron 1991, 7, 499–508. [Google Scholar] [CrossRef]
- Clerk, A.; Morris, G.E.; Dubowitz, V.; Davies, K.E.; Sewry, C.A. Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem. J. 1993, 25, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Miura, P.; Jasmin, B.J. Utrophin upregulation for treating duchenne or becker muscular dystrophy: How close are we? Trends Mol. Med. 2006, 12, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Cerletti, M.; Negri, T.; Cozzi, F.; Colpo, R.; Andreetta, F.; Croci, D.; Davies, K.E.; Cornelio, F.; Pozza, O.; Karpati, G.; et al. Dystrophic phenotype of canine x-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther. 2003, 10, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Tinsley, J.M.; Phelps, S.R.; Squire, S.E.; Townsend, E.R.; Martin, J.E.; Davies, K.E. Non-toxic ubiquitous over-expression of utrophin in the MDX mouse. Neuromuscul. Disord. 2001, 11, 713–721. [Google Scholar] [CrossRef]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- t Hoen, P.A.; de Meijer, E.J.; Boer, J.M.; Vossen, R.H.; Turk, R.; Maatman, R.G.; Davies, K.E.; van Ommen, G.J.; van Deutekom, J.C.; den Dunnen, J.T. Generation and characterization of transgenic mice with the full-length human DMD gene. J. Biol. Chem. 2008, 283, 5899–5907. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Ryu, S.-M.; Kim, S.-T.; Baek, G.; Kim, D.; Lim, K.; Chung, E.; Kim, S.; Kim, J.-S. Highly efficient RNA-guided base editing in mouse embryos. Nat. Biotechnol. 2017, 35, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, G.L.; Arechavala-Gomeza, V.; Fernandez-Fuente, M.; Burke, M.M.; Nagel, N.; Holder, A.; Stanley, R.; Chandler, K.; Marks, S.L.; Muntoni, F.; et al. A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skipping. PLoS ONE 2010, 5, e8647. [Google Scholar] [CrossRef] [PubMed]
- Hildyard, J.C.W.; Taylor-Brown, F.; Massey, C.; Wells, D.J.; Piercy, R.J. Determination of qpcr reference genes suitable for normalizing gene expression in a canine model of duchenne muscular dystrophy. J. Neuromuscul. Dis. 2018, 5, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Fujii, W.; Tsuboi, M.; Tanihata, J.; Teramoto, N.; Takeuchi, S.; Naito, K.; Yamanouchi, K.; Nishihara, M. Generation of muscular dystrophy model rats with a CRISPR/cas system. Sci. Rep. 2014, 4, 5635. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.H.; Zhao, H.; Qing, Y.B.; Pan, W.R.; Jia, B.Y.; Zhao, H.Y.; Huang, X.X.; Wei, H.J. Porcine zygote injection with Cas9/sgRNA results in DMD-modified pig with muscle dystrophy. Int. J. Mol. Sci. 2016, 17, 1668. [Google Scholar] [CrossRef] [PubMed]
- Sui, T.; Lau, Y.S.; Liu, D.; Liu, T.; Xu, L.; Gao, Y.; Lai, L.; Li, Z.; Han, R. A novel rabbit model of duchenne muscular dystrophy generated by CRISPR/Cas9. Dis. Model. Mech. 2018, 11, dmm032201. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, Y.; Kang, Y.; Yang, W.; Niu, Y.; Guo, X.; Tu, Z.; Si, C.; Wang, H.; Xing, R.; et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum. Mol. Genet. 2015, 24, 3764–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimo, T.; Hosoki, K.; Nakatsuji, Y.; Yokota, T.; Obika, S. A novel human muscle cell model of duchenne muscular dystrophy created by CRISPR/Cas9 and evaluation of antisense-mediated exon skipping. J. Hum. Genet. 2018, 63, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Yokota, T. Creation of DMD muscle cell model using CRISPR-Cas9 genome editing to test the efficacy of antisense-mediated exon skipping. Methods Mol. Biol. 2018, 1828, 165–171. [Google Scholar] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Crispr off-targets: A reassessment. Nat. Methods 2018, 15, 229–230. [CrossRef]
- Anderson, K.R.; Haeussler, M.; Watanabe, C.; Janakiraman, V.; Lund, J.; Modrusan, Z.; Stinson, J.; Bei, Q.; Buechler, A.; Yu, C.; et al. Crispr off-target analysis in genetically engineered rats and mice. Nat. Methods 2018, 15, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.E.; Gillum, D.; Kiani, S. Approaches to reduce CRISPR off-target effects for safer genome editing. Appl. Biosaf. 2017, 22, 7–13. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.R.; et al. Engineered CRISPR-Cas9 nucleases with altered pam specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.E.; Taussig, D.; Steinfeld, I.; Phadnis, S.M.; Lunstad, B.D.; Singh, M.; Vuong, X.; Okochi, K.D.; McCaffrey, R.; Olesiak, M.; et al. Improving CRISPR-Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res. 2018, 46, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. P53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. Crispr–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.L. Immunity to CRISPR Cas9 and Cas12a therapeutics. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1408. [Google Scholar] [CrossRef] [PubMed]
- Crudele, J.M.; Chamberlain, J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 2018, 9, 3497. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.L.; Amini, L.; Wendering, D.J.; Burkhardt, L.-M.; Akyüz, L.; Reinke, P.; Volk, H.-D.; Schmueck-Henneresse, M. High prevalence of streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- DiCarlo, J.E.; Deeconda, A.; Tsang, S.H. Viral vectors, engineered cells and the CRISPR revolution. Adv. Exp. Med. Biol. 2017, 1016, 3–27. [Google Scholar] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cas9 Enzyme | Source Bacteria | PAM Site | Protein, Gene Size | Key Features | Reference(s) |
|---|---|---|---|---|---|
| SpCas9 | Streptococcus pyogenes | 5′-NGG-3′ | 1368 aa, 4.10 kbp | Ubiquitous PAM site, widely used with multiples derivatives | [27,28,29] |
| SaCas9 | Staphylococcus aureus | 5′-NNGRRT-3′ | 1053 aa, 3.16 kbp | Smaller size, better packaged for viral delivery | [30,31] |
| CjCas9 | Campylobacter jejuni | 5′-NNNNACAC-3′, 5′-NNNNRYAC-3′ | 984 aa, 2.95 kbp | Even smaller than SaCas9, lower chance of off-targeting due to longer PAM | [32] |
| Cas Enzyme | Strategy | Target Gene Region(s) | Model(s) | Delivery | Study Highlights | Reference |
|---|---|---|---|---|---|---|
| SpCas9 | NHEJ reframing, HDR exon correction | Dmd exon 23 | mdx mice | 1-cell embryo injection | Dystrophin restoration observed by IHC (up to 100%) and WB; 17% Dmd HDR correction resulted in 47–60% dystrophin-positive fibers in skeletal muscles and the heart | 2014 Long et al. [43] |
| SpCas9 | NHEJ reframing, exon skipping, HDR exon knock-in | DMD intron 44/exon 45 | DMD hiPSCs, hiPSC-derived skeletal muscle cells (ex44 del.) | Electroporation | Dystrophin restoration in derived skeletal muscle cells observed by WB and IHC for all strategies; CRISPR was as effective as using TALEN | 2015 Li et al. [41] |
| SpCas9 | NHEJ reframing, single/multiple exon deletion | DMD exons 45–55 (for reframing each exon), introns 50 and 51 (ex51 del.), introns 44 and 55 (ex45–55 del.) | immortalized DMD patient muscle cells (ex48–50 del.), immunodeficient NSG mice | Electroporation | Generated targeted deletions of exon/s in vitro, particularly of the large exon 45–55 region which led to dystrophin rescue by WB; mice transplanted with treated myoblasts (exon 51-deleted) showed dystrophin-positive fibers by IHC | 2015 Ousterout et al. [21] |
| dSpCas9-VP16 | Utrophin upregulation | UTRN A/B promoter | immortalized DMD patient muscle cells (ex45–52 del.) | Electroporation | 1.7–6.9-fold upregulation of utrophin achieved; restored β-dystroglycan expression observed by WB with as little as 1.7-fold upregulation | 2016 Wojtal et al. [44] |
| SpCas9 | Duplicated exons removal | DMD intron 27 | primary DMD patient fibroblasts (ex18–30 dup.) | LV transduction, with Adeno-MyoD | 4.42% full-length dystrophin production achieved post-treatment, accompanied with α-dystroglycan restoration | 2016 Wojtal et al. [44] |
| SpCas9 | Single exon deletion | Dmd exon 23, introns 22 and 23 (ex23 del.) | mdx mice | AAV9 delivery (i.m., i.p., i.v.) | All modes of injection led to appearance of dystrophin-positive fibers as evaluated by IHC: ~25.5% 6 wks post-i.m., ~4.6% and ~9.6% in skeletal and cardiac muscles respectively 12 wks post-i.v., ~1.8% and ~3.2% in skeletal and cardiac muscles respectively 8 wks post-i.p. | 2016 Long et al. [45] |
| SaCas9 | Single exon deletion | Dmd introns 22 and 23 (ex23 del.) | mdx mice | AAV8 delivery (i.m., i.p., i.v.) | Intramuscular injections led to ~59% of transcripts with exon 23 deleted, which restored about 8% dystrophin of healthy levels by WB, proper relocalization of DGC proteins, and muscle function improvement; systemic injections restored dystrophin production in the heart and skeletal muscles | 2016 Nelson et al. [46] |
| SpCas9, SaCas9 | Single exon deletion | Dmd introns 22 and 23 (ex23 del.) | mdx mice, mdx satellite cells | AAV9 delivery (i.m., i.p., i.v.) | Dual-vector (Cas9 and gRNAs on separate constructs) had higher cutting efficiency than a single-vector system (Cas9 and gRNAs on the same construct) in vitro; dystrophin restoration >10% observed in the heart and skeletal muscles upon systemic treatment; correction also possible in satellite cells | 2016 Tabebordbar et al. [47] |
| SpCas9 | Hybrid exon formation via internal exon deletion | DMD exons 50 and 54 | immortalized DMD patient muscle cells (ex51–53 del.), hDMD/mdx mice | Lipotransfection (in vitro)/ electroporation (in vivo) | Dystrophin restoration successful in vitro by WB, not shown in vivo; hybrid exon formation thought to preserve dystrophin rod domain structure better | 2016 Iyombe-Engembe et al. [48] |
| SpCas9 | NHEJ reframing, single/multiple exon deletion | DMD exons 51, 53, introns 52 and 53 (ex53 del.), 43 and 54 (ex44–54 del.) | immortalized DMD patient muscle cells (ex48–50, or 45–52 del.) | Sequential LV then AdV transduction/AdV transduction | Study showed the possibility of combining both TALEN and CRISPR approaches in one gene editing strategy; also, comparable editing was obtained with Cas9 and gRNA delivered either together or separately in AdV | 2016 Maggio et al. [49] |
| SpCas9 | Multiple exon deletion | Dmd introns 20 and 23 (ex21–23 del.) | mdx mice | Electroporation/AdV transduction | Treatment restored proper calcium dynamics in muscle (electroporation), and restored dystrophin to 50% of wild-type levels, as well as dystrophin-associated complex sarcolemmal localization and muscle membrane integrity (transduction) | 2016 Xu et al. [50] |
| SpCas9 | Multiple exon deletion | DMD introns 44 and 55 (ex45–55 del.) | DMD hiPSCs, hiPSC-derived skeletal and cardiac muscle cells (ex46–51 or 46–47 del., ex50 dup.), immunodeficient NSG-mdx mice | Nucleofection | CRISPR-mediated deletion of the large exon 45–55 region achieved, restored membrane function and dystrophin, β-dystroglycan expression by WB and IHC; mice transplanted with hiPSC-derived skeletal muscle cells showed dystrophin-positive fibers by IHC | 2016 Young et al. [42] |
| SpCas9 | NHEJ reframing, single/multiple exon deletion | DMD exons 51, 53 (for reframing) introns 52 and 53 (ex53 del.), introns 43 and 54 (ex44–54 del.) | immortalized DMD patient muscle cells (ex48–50, or 45–52 del.) | AdV transduction | AdV with 2gRNA-SpCas9 constructs work as good as those with 1gRNA-SpCas9 constructs in terms of corrective ability and dystrophin restoration | 2016 Maggio et al. [51] |
| SpCas9, SaCas9 | Multiple exon deletion, HDR exon correction | Dmd exon 53, introns 51 and 53 (ex52–53 del.) | mdx4cv mice (nonsense ex53 mutation) | AAV6 delivery (i.m., i.v.) | Dual vector approach (SpCas9 and gRNA separate) yielded higher correction efficiency than single vector approach (SaCas9 and gRNA together); systemic treatment restored dystrophin expression in the heart (~34% dystrophin-positive fibers) and skeletal muscles (~10–50% dystrophin-positive fibers) | 2017 Bengtsson et al. [52] |
| LbCpf1, AsCpf1 | NHEJ reframing, single exon skipping, HDR exon correction | DMD exon 51, intron 50 | DMD hiPSCs, hiPSC-derived cardiac muscle cells (ex48–50 del.), mdx mice | Nucleofection (in vitro)/ 1-cell embryo injection (in vivo) | Cpf1 editing successfully restored dystrophin expression and improved mitochondrial function in cardiomyocytes; 5/24 pups (injected at the embryo stage) showed HDR correction and had ameliorated dystrophic phenotypes | 2017 Zhang et al. [53] |
| SpCas9 | Duplicated exon removal | DMD exon 2, intron 2 | immortalized DMD patient muscle cells (ex2 dup.) | PEI transfection/LV transduction | Use of a single gRNA can delete a duplicated exon, resulting in slight dystrophin rescue by WB and IHC | 2017 Lattanzi et al. [54] |
| SpCas9 | HDR exon correction | Dmd exon 23 | mdx mice, mdx satellite cells | Lipotransfection (template, gRNA), AdV transduction (Cas9)/AdV transduction | Higher transduction efficiency obtained when AdVs were used for both Cas9 and gRNA-HDR template delivery; mice transplanted with corrected satellite cells showed dystrophin-positive fibers by IHC | 2017 Zhu et al. [55] |
| SpCas9 | Multiple exon deletion | DMD introns 44 and 55 (ex45–55 del.) | humanized mdx mice with DMD exon 45 del. | Electroporation | Exon 45–55 deletion by CRISPR possible in vivo; first use of the humanized DMD mouse model with exon 45 del. for CRISPR studies | 2017 Young et al. [56] |
| SpCas9 | Multiple exon deletion | DMD introns 2 and 7 (ex3–9 del.), introns 5 and 7 (ex6–7 del.), introns 6 and 11 (ex7–11 del.) | DMD hiPSCs, hiPSC-derived cardiac muscle cells (ex8–9 or ex3–7 del.) | Nucleofection | Dystrophin with ex7–11 del. showed the least functionality, while those with ex3–9 del. had the highest functionality in terms of assessing iPSC-derived cardiomyocyte calcium cycling | 2017 Kyrychenko et al. [57] |
| SpCas9 | HDR correction | Dmd exon 23 | mdx primary muscle cells, mdx mice | CRISPR-Gold nanoparticles (i.m.) | 5.4% HDR correction of the Dmd mutation in mdx was observed after CRISPR treatment and cardiotoxin injection, dystrophin-positive fibers found by IHC; 0.8% HDR correction observed without cardiotoxin co-injection, which led to significantly improved hanging test performance | 2017 Lee et al. [58] |
| SpCas9 | NHEJ reframing, single exon skipping | Dmd exon 51 | mice with Dmd exon 50 del. | AAV9 delivery (i.m., i.p.) | Successful dystrophin restoration in the heart and skeletal muscles; systemic injections led to improved muscle function; first application of CRISPR in the ex50 del. mouse model | 2017 Amoasii et al. [59] |
| SpCas9 | Single exon deletion | Dmd introns 50 and 51 (ex51 del.) | primary human skeletal muscle cells | HCAdV delivery | Up to 93.3% exon 51 deletion observed in vitro upon delivery of CRISPR agents by HCAdV | 2017 Ehrke-Schulz et al. [60] |
| SpCas9 | NHEJ reframing, exon skipping | DMD exon 51, introns 47, 50, 54 | DMD hiPSCs, hiPSC-derived cardiac muscle cells (ex48–50 del., pseudo-ex47, ex55–59 dup.) | Nucleofection | All strategies corrected the respective patient mutations and restored dystrophin production in iPSC-derived cardiomyocytes; 3D-engineered heart muscle produced from treated iPSC-derived cardiomyocytes showed improved contractile force | 2018 Long et al. [61] |
| CjCas9 | NHEJ reframing | Dmd exon 23 | mice with deletions in Dmd exon 23 | AAV9 delivery (i.m.) | CjCas9 displayed higher targeting specificity than SpCas9; use of CjCas9-based CRISPR can lead to successful dystrophin restoration and improvement in muscle function as well | 2018 Koo et al. [62] |
| SaCas9 | Hybrid exon formation via multiple exon deletion | DMD exons 47 and 58 | DMD skeletal muscle cells (ex51–53 del., ex49–50 del., ex51–56 del., ex50–52 del.), humanized mdx mice with DMD ex52 del. | LV transduction (in vitro)/AAV9 delivery (in vivo; i.v.) | gRNAs designed to produce exon deletions that best preserved dystrophin protein structure were able to show dystrophin restoration in vitro and in vivo (slight rescue in the heart) | 2018 Duchêne et al. [63] |
| SpCas9 | NHEJ reframing, exon skipping | Dystrophin exon 51 | deltaE50-MD canine model (ex50 del.) | AAV9 delivery (i.m., i.v.) | First published study on dystrophin gene correction in a dog model; ~3–70% dystrophin restoration of healthy levels in skeletal muscles and ~92% in the heart found by WB | 2018 Amoasii et al. [64] |
| nSpCas9-ABE7.10 | Base editing to correct a nonsense mutation | Dmd exon 20 | mice with a nonsense mutation in Dmd exon 20 | trans-splicing AAV2/9 delivery (i.m.) | ~3.3% base editing frequency achieved 8 weeks post-treatment with no detectable off-target effects; ~17% dystrophin-positive fibers and restored localization of nNOS observed by IHC | 2018 Ryu et al. [65] |
| dSa/SpCas9-TAM | Base editing to induce exon skipping | DMD intron 50 5′ splice site | DMD hiPSCs, hiPSC-derived cardiac muscle cells (ex51 del.) | Lipotransfection | ~100% base editing efficiency achieved; corrected iPSC-derived cardiomyocytes had restored dystrophin protein, low CK and miR-31 levels, and restoration of β-dystroglycan expression | 2018 Yuan et al. [66] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, K.R.Q.; Yoon, C.; Yokota, T. Applications of CRISPR/Cas9 for the Treatment of Duchenne Muscular Dystrophy. J. Pers. Med. 2018, 8, 38. https://doi.org/10.3390/jpm8040038
Lim KRQ, Yoon C, Yokota T. Applications of CRISPR/Cas9 for the Treatment of Duchenne Muscular Dystrophy. Journal of Personalized Medicine. 2018; 8(4):38. https://doi.org/10.3390/jpm8040038
Chicago/Turabian StyleLim, Kenji Rowel Q., Chantal Yoon, and Toshifumi Yokota. 2018. "Applications of CRISPR/Cas9 for the Treatment of Duchenne Muscular Dystrophy" Journal of Personalized Medicine 8, no. 4: 38. https://doi.org/10.3390/jpm8040038