
Genetic Screening Reveals Heterogeneous Clinical Phenotypes in Patients with Dilated Cardiomyopathy and Troponin T2 Variants
 , and
, and
Abstract
:1. Background
Troponin T2, Cardiac Type (TNNT2)
2. Materials and Methods
2.1. Patients
2.2. Cardiac Histology
3. Results
3.1. Family A
3.2. Family B
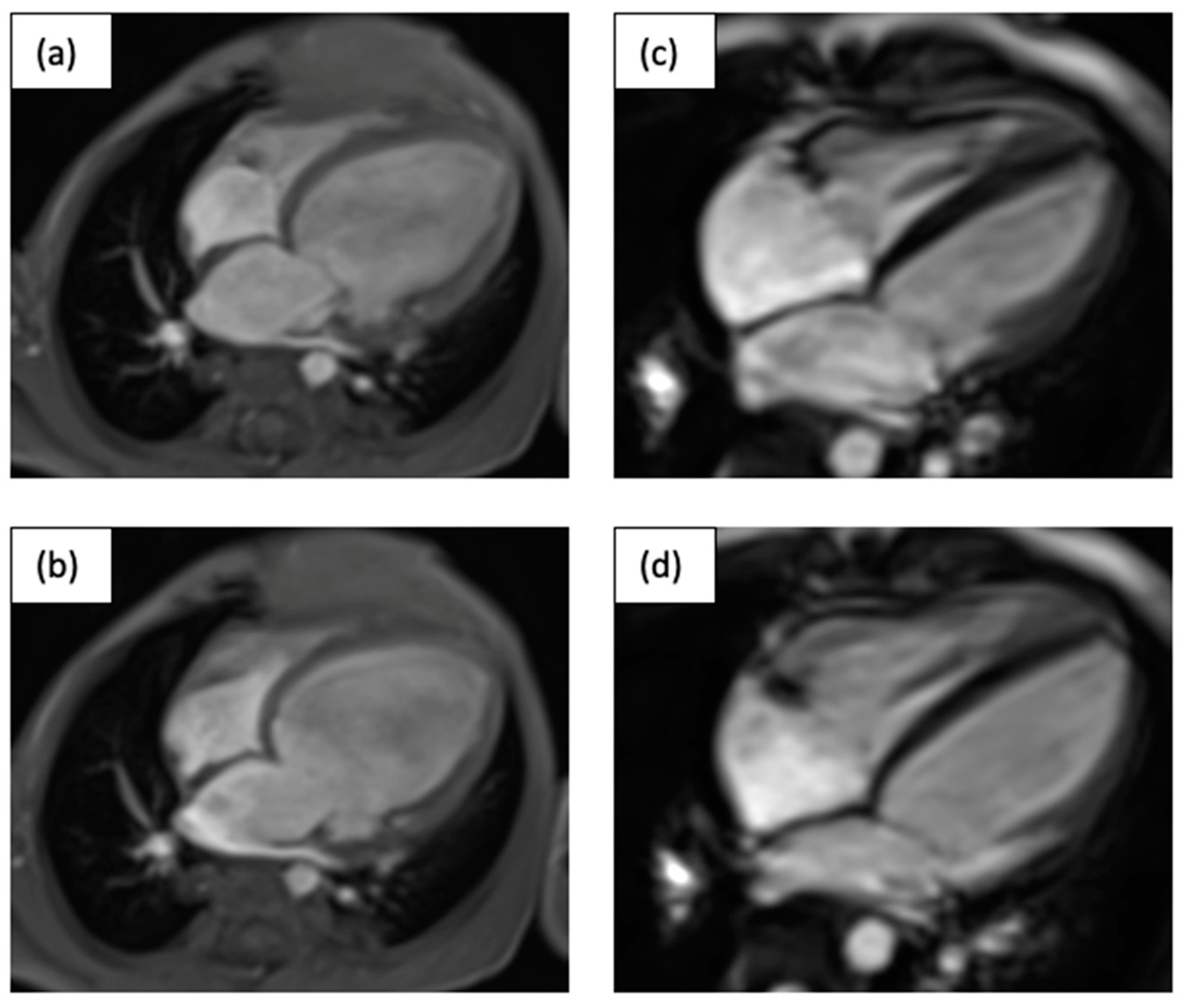
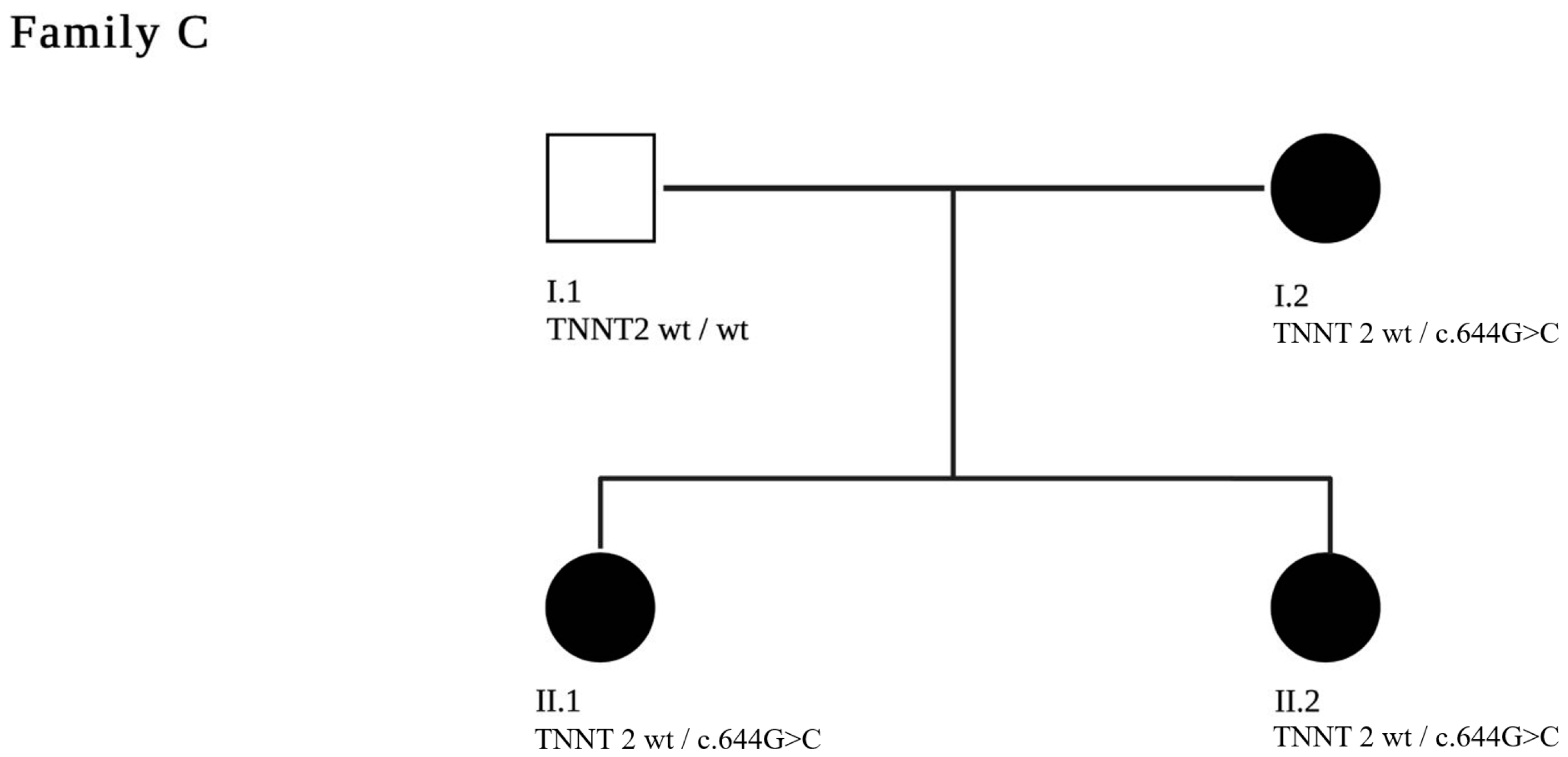
3.3. Family C
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation 2019, 140, e9–e68. [Google Scholar] [CrossRef] [PubMed]
- Zschirnt, M.; Thul, J.; Akintürk, H.; Valeske, K.; Schranz, D.; Skrzypek, S.; Müller, M.; Jux, C.; Hahn, A.; Rupp, S. Aetiology and 30-Year Long-Term Outcome of Children with Cardiomyopathy Necessitating Heart Transplantation. J. Pers. Med. 2020, 10, 251. [Google Scholar] [CrossRef] [PubMed]
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated cardiomyopathy: From epidemiologic to genetic phenotypes: A translational review of current literature. J. Intern. Med. 2019, 286, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, B.; Jin, J.P. TNNT1, TNNT2, and TNNT3: Isoform genes, regulation, and structure-function relationships. Gene 2016, 582, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnall, R.D.; Singer, E.S.; Wacker, J.; Nowak, N.; Ingles, J.; King, I.; Macciocca, I.; Crowe, J.; Ronan, A.; Weintraub, R.G.; et al. Genetic Basis of Childhood Cardiomyopathy. Circ. Genom. Precis Med. 2022, 15, e003686. [Google Scholar] [CrossRef] [PubMed]
- Vadgama, N.; Ameen, M.; Sundaram, L.; Gaddam, S.; Gifford, C.; Nasir, J.; Karakikes, I. Genomics England Research Consortium De novo and inherited variants in coding and regulatory regions in genetic cardiomyopathies. Hum. Genom. 2022, 16, 55. [Google Scholar] [CrossRef] [PubMed]
- Webber, S.A.; Lipshultz, S.E.; Sleeper, L.A.; Lu, M.; Wilkinson, J.D.; Addonizio, L.J.; Canter, C.E.; Colan, S.D.; Everitt, M.D.; Jefferies, J.L.; et al. Pediatric Cardiomyopathy Registry, Outcomes of restrictive cardiomyopathy in childhood and the influence of phenotype: A report from the Pediatric Cardiomyopathy Registry. Circulation 2012, 126, 1237–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, M.; Ehlermann, P.; Weichenhan, D.; Will, R.; Zeller, R.; Rupp, S.; Müller, A.; Steen, H.; Ivandic, B.T.; Ulmer, H.E.; et al. Severe familial left ventricular non-compaction cardiomyopathy due to a novel troponin T (TNNT2) mutation. Cardiovasc. Res. 2010, 86, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogensen, J.; Murphy, R.T.; Shaw, T.; Bahl, A.; Redwood, C.; Watkins, H.; Burke, M.; Elliott, P.M.; McKenna, W.J. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 2033–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatraman, G.; Gomes, A.V.; Glenn, W.; Kerrick, L.; Potter, J.D. Characterization of troponin T dilated cardiomyopathy mutations in the fetal troponin isoform. J. Biol. Chem. 2005, 280, 17584–17592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, R.L.; French, K.S.; Resta, R.G.; Doyle, D.L. Standardized human pedigree nomenclature: Update and assessment of the recommendations of the National Society of Genetic Counselors. J. Genet. Couns. 2008, 17, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Committee, Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schranz, D.; Akintuerk, H.; Bailey, L. Pulmonary Artery Banding for Functional Regeneration of End-Stage Dilated Cardiomyopathy in Young Children: World Network Report. Circulation 2018, 137, 1410–1412. [Google Scholar] [CrossRef] [PubMed]
- Dellefave, L.; McNally, E.M. Sarcomere mutations in cardiomyopathy, noncompaction, and the developing heart. Circulation 2008, 117, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Seidel, F.; Holtgrewe, M.; Al-Wakeel-Marquard, N.; Opgen-Rhein, B.; Dartsch, J.; Herbst, C.; Beule, D.; Pickardt, T.; Klingel, K.; Messroghli, D.; et al. Pathogenic Variants Associated With Dilated Cardiomyopathy Predict Outcome in Pediatric Myocarditis. Circ. Genom. Precis Med. 2021, 14, e003250. [Google Scholar] [CrossRef] [PubMed]
- Mandawat, A.; Chattranukulchai, P.; Mandawat, A.; Blood, A.J.; Ambati, S.; Hayes, B.; Rehwald, W.; Kim, H.W.; Heitner, J.F.; Shah, D.J.; et al. Progression of Myocardial Fibrosis in Nonischemic DCM and Association with Mortality and Heart Failure Outcomes. JACC Cardiovasc. Imaging 2021, 14, 1338–1350. [Google Scholar] [CrossRef] [PubMed]
- Nicin, L.; Abplanalp, W.T.; Schanzer, A.; Sprengel, A.; John, D.; Mellentin, H.; Tombor, L.; Keuper, M.; Ullrich, E.; Klingel, K.; et al. Single Nuclei Sequencing Reveals Novel Insights Into the Regulation of Cellular Signatures in Children with Dilated Cardiomyopathy. Circulation 2021, 143, 1704–1719. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Admission | Follow up at Last Contact, #, & | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gender | Age | LVEDD (z-Score) | EF (%) | BNP (pg/mL) | Age | LVEDD (z-Score) | EF (%) | BNP (pg/mL) | |
| A.III.3 | female | 2 y | +7.2 | severely reduced | 6528 | # at the age of 2 y | # | # | # |
| A.III.2 | male | 0 m | +2.2 | normal | 206 | # at the age of 13 y | # | # | # |
| A.III.1 | male | 6 m | −0.9 | 64 | n.a. | 5 y | +3.9 | 66 | n.a. |
| B. II.3 | male | n.a. | n.a. | n.a. | n.a. | & at the age 50 y | & | & | & |
| B.III.3 | male | 5 m | +8.4 | reduced | n.a. | # at the age of 8 m, & at the age of 12 y | & | & | & |
| B.III.4 | male | 12 y | 7.7 | severely reduced | n.a. | & at the age 12 y | & | & | & |
| B.III.5 | female | 10 y | 1.5 | mildly reduced | n.a. | # at the age of 14 y, & at the age of 20 y | & | & | & |
| B.III.6 | male | 21 m | −0.4 | normal | n.a. | 12 y | +1.0 | 51 | <50 |
| C.II.1 | female | 4 y | +4.6 | 25 | 3651 | 12 y | +4.6 | 48 | 59 |
| C.II.2 | female | 14 y | +5.1 | 62 | n.a. | 16 y | +4.8 | 48 | 29 |
| C.I.2 | female | 35 y | n.a. | n.a. | n.a. | 39 y | +5.2 | n.a. | n.a. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weis, A.; Krueck, S.; Dombrowsky, G.; Schänzer, A.; Jux, C.; Uebing, A.; Voges, I.; Hitz, M.-P.; Rupp, S. Genetic Screening Reveals Heterogeneous Clinical Phenotypes in Patients with Dilated Cardiomyopathy and Troponin T2 Variants. J. Pers. Med. 2023, 13, 611. https://doi.org/10.3390/jpm13040611
Weis A, Krueck S, Dombrowsky G, Schänzer A, Jux C, Uebing A, Voges I, Hitz M-P, Rupp S. Genetic Screening Reveals Heterogeneous Clinical Phenotypes in Patients with Dilated Cardiomyopathy and Troponin T2 Variants. Journal of Personalized Medicine. 2023; 13(4):611. https://doi.org/10.3390/jpm13040611
Chicago/Turabian StyleWeis, Angelika, Svenja Krueck, Gregor Dombrowsky, Anne Schänzer, Christian Jux, Anselm Uebing, Inga Voges, Marc-Phillip Hitz, and Stefan Rupp. 2023. "Genetic Screening Reveals Heterogeneous Clinical Phenotypes in Patients with Dilated Cardiomyopathy and Troponin T2 Variants" Journal of Personalized Medicine 13, no. 4: 611. https://doi.org/10.3390/jpm13040611