
Pharmacogenetics and Adverse Events in the Use of Fluoropyrimidine in a Cohort of Cancer Patients on Standard of Care Treatment in Zimbabwe

 , , ,
, , ,
Abstract
:1. Introduction
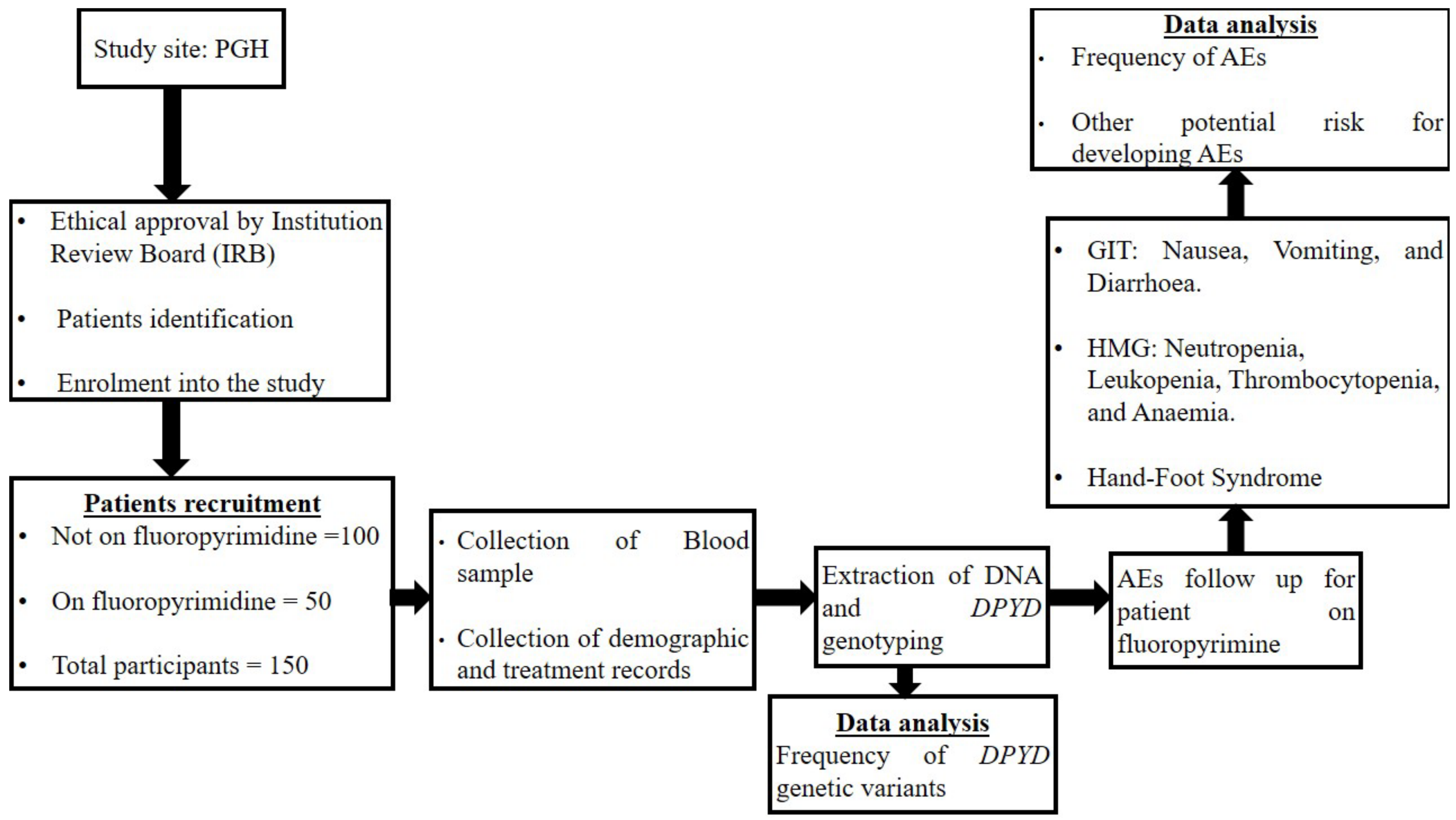
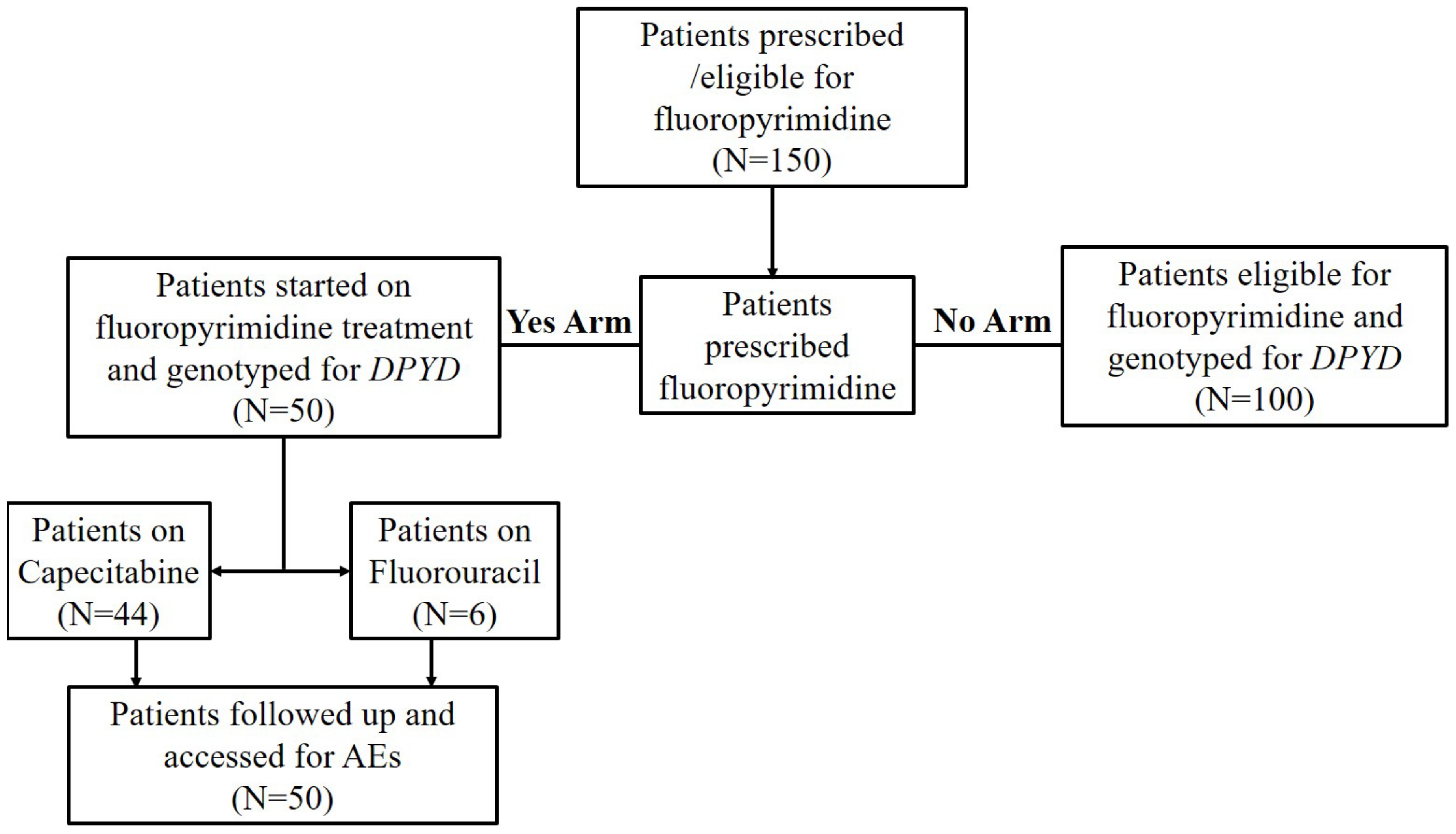
2. Materials and Methods
3. Results
3.1. Study Participants Characteristics
3.2. Allele Frequency of DPYD Pathogenic Variants in the Guidelines
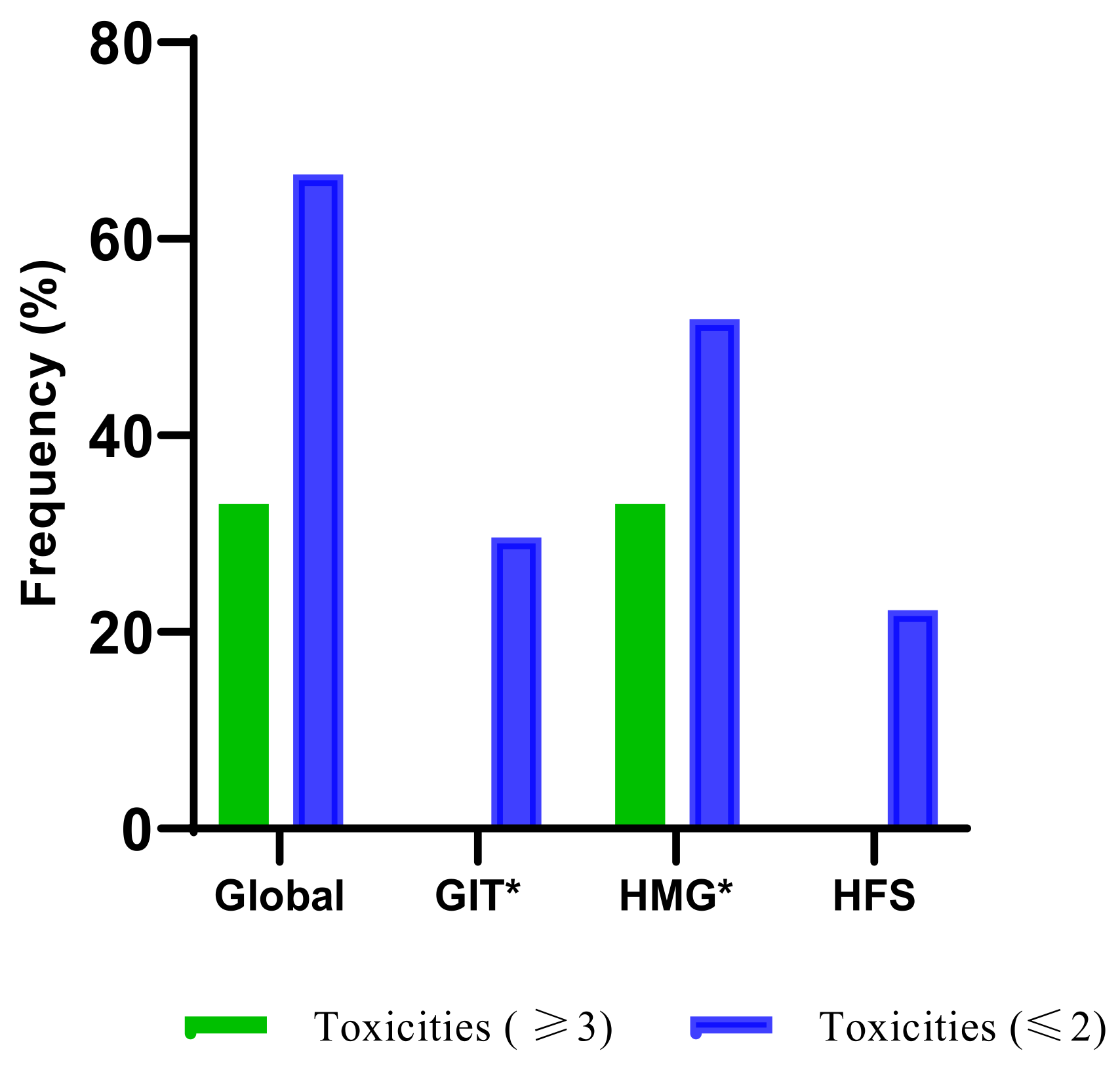
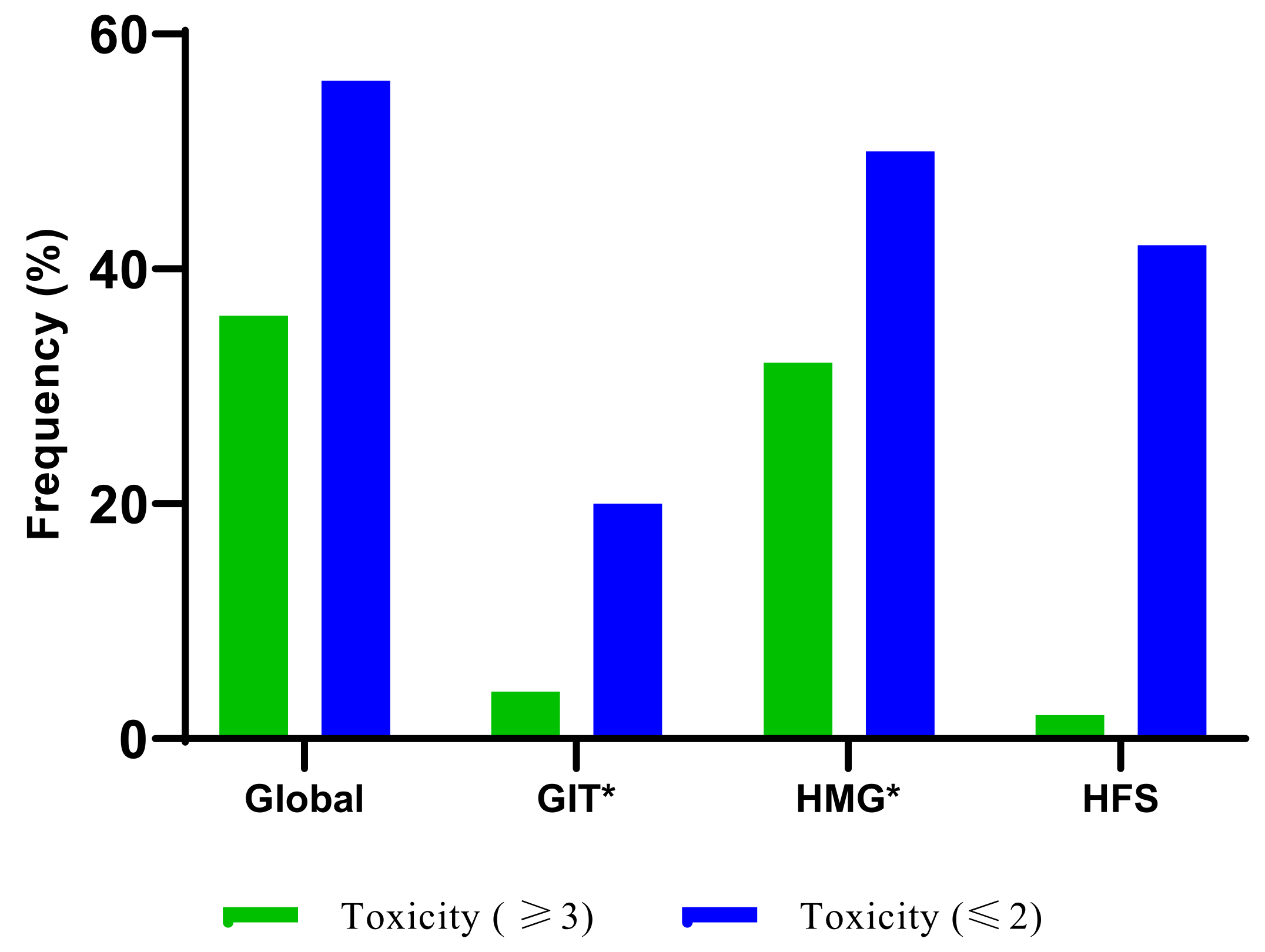
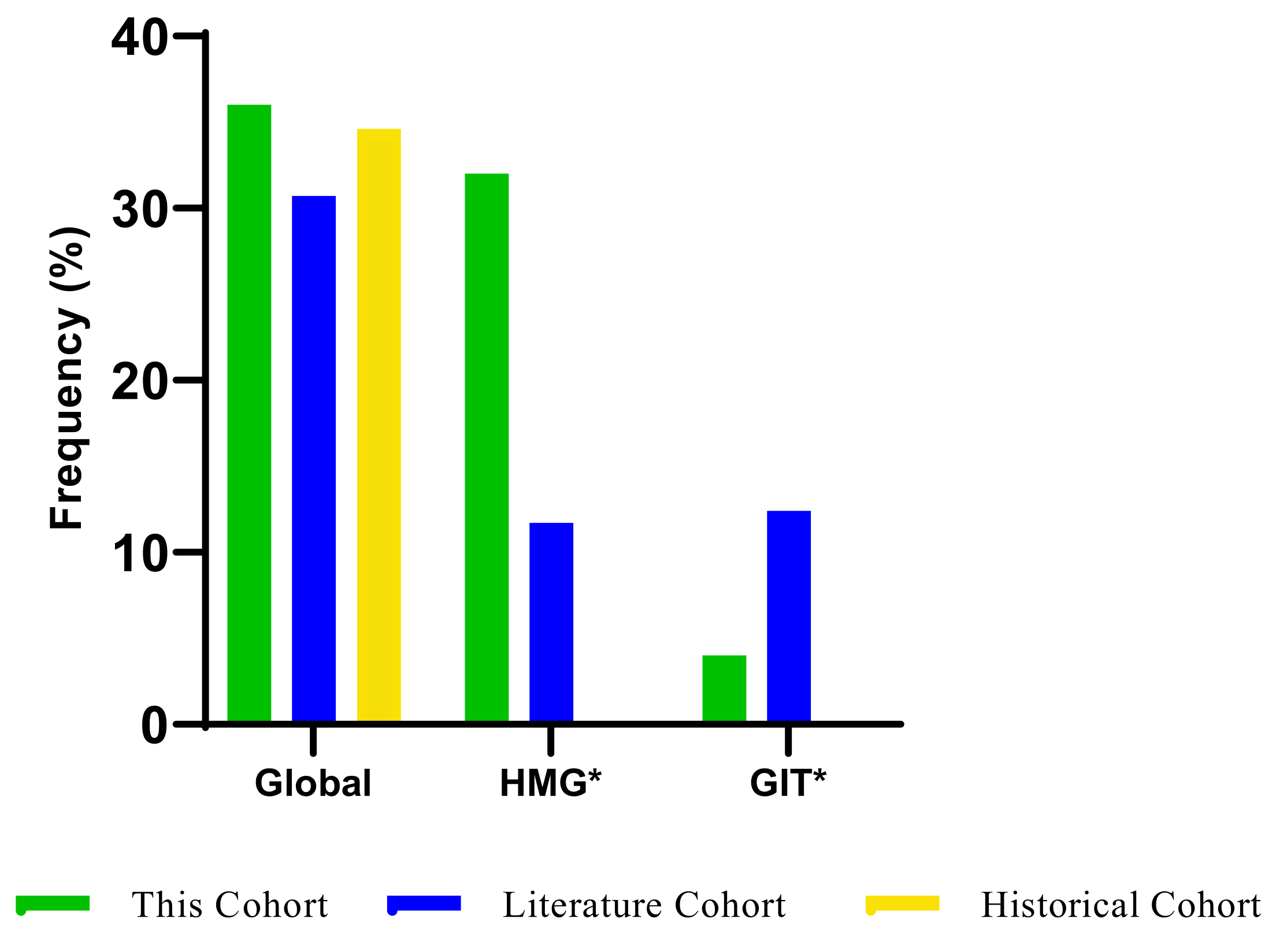
3.3. The Frequency of Fluoropyrimidine-Related AEs
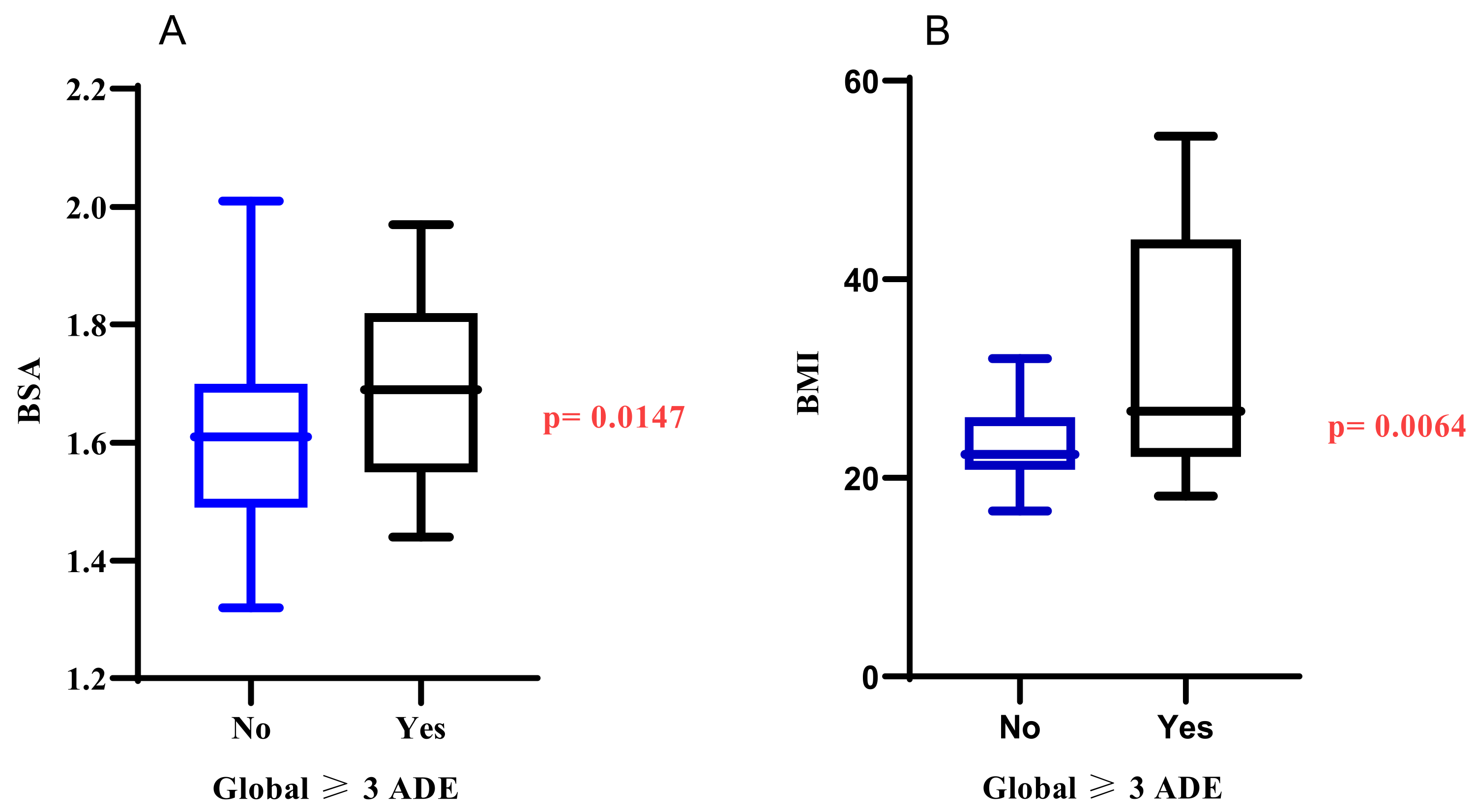
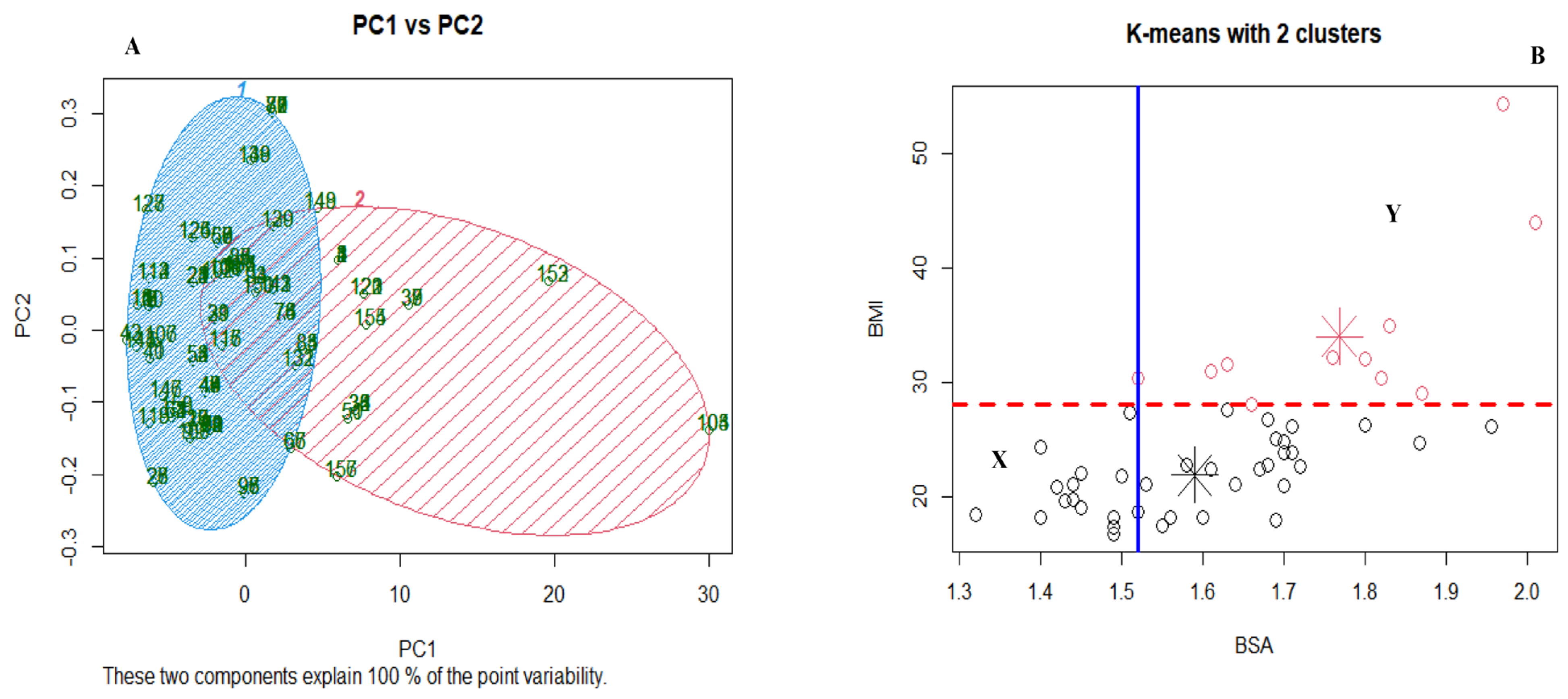
3.4. Other Potential Risks of Developing AEs Using Gastrointestinal and Haematological Adverse Events as Surrogate Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dean, L.; Fluorouracil Therapy and DPYD Genotype. Medical Genetics Summaries. 2012. Available online: http://www.ncbi.nlm.nih.gov/pubmed/28520376 (accessed on 24 November 2022).
- Casale, J.; Patel, P. Fluorouracil; StatPearls Publishing: Treasure Island, FL, USA, 2022; pp. 6–8. [Google Scholar]
- Innocenti, F.; Mills, S.C.; Sanoff, H.; Ciccolini, J.; Lenz, H.-J.; Milano, G. All You Need to Know About DPYD Genetic Testing for Patients Treated with Fluorouracil and Capecitabine: A Practitioner-Friendly Guide. JCO Oncol. Pract. 2020, 16, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, F. DPYD variants to predict 5-FU toxicity: The ultimate proof. J. Natl. Cancer Inst. 2014, 106, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Wigle, T.J.; Tsvetkova, E.V.; Welch, S.A.; Kim, R.B. DPYD and fluorouracil-based chemotherapy: Mini review and case report. Pharmaceutics 2019, 11, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varughese, L.A.; Lau-Min, K.S.; Cambareri, C.; Damjanov, N.; Massa, R.; Reddy, N.; Oyer, R.; Teitelbaum, U.; Tuteja, S. DPYD and UGT1A1 Pharmacogenetic Testing in Patients with Gastrointestinal Malignancies: An Overview of the Evidence and Considerations for Clinical Implementation. Pharmacotherapy 2020, 40, 1108–1129. [Google Scholar] [CrossRef] [PubMed]
- Seid, H.; Aebi, S.; Joerger, M.; Montemurro, M.; Ansari, M.; Amstutz, U.; Largiadèr, C. Fluoropyrimidine chemotherapy: Recommendations for DPYD genotyping and therapeutic drug monitoring of the Swiss Group of Pharmacogenomics and Personalised Therapy. Swiss Med. Wkly. 2020, 150, w20375. [Google Scholar] [CrossRef]
- Kim, S.R.; Park, C.H.; Park, S.; Park, J.O.; Lee, J.; Lee, S.Y. Genetic polymorphisms associated with 5-fluorouracil-induced neurotoxicity. Chemotherapy 2010, 56, 313–317. [Google Scholar] [CrossRef]
- Amstutz, U.; Henricks, L.; Offer, S.M.; Barbarino, J.; Schellens, J.H.; Swen, J.; Klein, T.E.; McLeod, H.L.; Caudle, K.E.; Diasio, R.B.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin. Pharmacol. Ther. 2018, 103, 210–216. [Google Scholar] [CrossRef]
- Deenen, M.J.; Meulendijks, D.; Cats, A.; Sechterberger, M.K.; Severens, J.L.; Boot, H.; Smits, P.H.; Rosing, H.; Mandigers, C.M.; Soesan, M. Upfront genotyping of DPYD* 2A to individualize fluoropyrimidine therapy: A safety and cost analysis. J. Clin. Oncol. 2016, 34, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Lin, Y.; Zou, B.; Liu, W.; Zhang, Y.; Zhao, L.; Huang, Y.; Yang, Y.; Fang, W.; Zhao, Y.; et al. Pharmacokinetic and Pharmacodynamic Analyses of 5-Fluorouracil in East-Asian Patients with Nasopharyngeal Carcinoma. Clin. Pharmacokinet. 2016, 55, 1205–1216. [Google Scholar] [CrossRef]
- Lunenburg, C.A.T.C.; Henricks, L.M.; Guchelaar, H.-J.; Swen, J.J.; Deenen, M.J.; Schellens, J.H.; Gelderblom, H. Prospective DPYD genotyping to reduce the risk of fluoropyrimidine-induced severe toxicity: Ready for prime time. Eur. J. Cancer 2016, 54, 40–48. [Google Scholar] [CrossRef]
- Henricks, L.M.; Lunenburg, C.A.T.C.; de Man, F.; Meulendijks, D.; Frederix, G.W.J.; Kienhuis, E.; Creemers, G.-J.; Baars, A.; Dezentjé, V.O.; Imholz, A.L.T.; et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: A prospective safety analysis. Lancet Oncol. 2018, 19, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Knikman, J.E.; Gelderblom, H.; Beijnen, J.H.; Cats, A.; Guchelaar, H.; Henricks, L.M. Individualized Dosing of Fluoropyrimidine-Based Chemotherapy to Prevent Severe Fluoropyrimidine-Related Toxicity: What Are the Options? Clin. Pharmacol. Ther. 2021, 109, 591–604. [Google Scholar] [CrossRef]
- Da Rocha, J.E.B.; Lombard, Z.; Ramsay, M. Potential Impact of DPYD Variation on Fluoropyrimidine Drug Response in sub-Saharan African Populations. Front. Genet. 2021, 12, 626954. [Google Scholar] [CrossRef]
- Alessandrini, M.; Chaudhry, M.; Dodgen, T.M.; Pepper, M.S. Pharmacogenomics and global precision medicine in the context of adverse drug reactions: Top 10 opportunities and challenges for the next decade. Omi. J. Integr. Biol. 2016, 20, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Abdullah-Koolmees, H.; Van Keulen, A.M.; Nijenhuis, M. Pharmacogenetics Guidelines: Overview and Comparison of the DPWG, CPIC, CPNDS, and RNPGx. Front. Pharmacol. 2021, 11, 595219. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-Y. The implementation of pharmacogenomics for personalized pharmacotherapy in Korea. Drug Metab. Pharmacokinet. 2017, 32, S16. [Google Scholar] [CrossRef]
- Lunenburg, C.A.T.C.; van der Wouden, C.H.; Nijenhuis, M.; Rhenen, M.H.C.-V.; de Boer-Veger, N.J.; Buunk, A.M.; Houwink, E.J.F.; Mulder, H.; Rongen, G.A.; van Schaik, R.H.N.; et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene–drug interaction of DPYD and fluoropyrimidines. Eur. J. Hum. Genet. 2020, 28, 508–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GLOBOCAN. Zimbabwe fact sheets. 14 862 927. 2020. Available online: https://gco.iarc.fr/today/data/factsheets/populations/716-zimbabwe-fact-sheets.pdf (accessed on 21 December 2022).
- GLOBOCAN. Africa fact sheets. 1 340 598 088. 2021. Available online: https://gco.iarc.fr/today/data/factsheets/populations/903-africa-fact-sheets.pdf (accessed on 18 February 2023).
- U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) v.5.0. Cancer Therapy Evaluation Program 2017. Available online: http://upen.terengganu.gov.my/index.php/2017 (accessed on 3 August 2022).
- Meulendijks, D.; Henricks, L.; Sonke, G.; Deenen, M.J.; Froehlich, T.K.; Amstutz, U.; Largiader, C.; Jennings, B.; Marinaki, A.M.; Sanderson, J.D.; et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: A systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015, 16, 1639–1650. [Google Scholar] [CrossRef]
- Wigle, T.J.; Povitz, B.L.; Medwid, S.; Teft, W.A.; Legan, R.M.; Lenehan, J.; Nevison, S.; Panuganty, V.; Keller, D.; Mailloux, J.; et al. Impact of pretreatment dihydropyrimidine dehydrogenase genotype-guided fluoropyrimidine dosing on chemotherapy associated adverse events. Clin. Transl. Sci. 2021, 14, 1338–1348. [Google Scholar] [CrossRef]
- Curigliano, G.; Banerjee, S.; Cervantes, A.; Garassino, M.C.; Garrido, P.; Girard, N.; Haanen, J.; Jordan, K.; Lordick, F.; Machiels, J.P.; et al. Managing cancer patients during the COVID-19 pandemic: An ESMO multidisciplinary expert consensus. Ann. Oncol. 2020, 31, 1320–1335. [Google Scholar] [CrossRef]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbavha, B.T.; Kanji, C.R.; Stadler, N.; Stingl, J.; Stanglmair, A.; Scholl, C.; Wekwete, W.; Masimirembwa, C. Population genetic polymorphisms of pharmacogenes in Zimbabwe, a potential guide for the safe and efficacious use of medicines in people of African ancestry. Pharmacogenet. Genom. 2022, 32, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, T.K.; Amstutz, U.; Aebi, S.; Joerger, M.; Largiade, C.R. Clinical importance of risk variants in the dehydrogenase gene for the prediction of early-onset fluoropyrimidine toxicity. Int. J. Cancer 2015, 136, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.D.; Bates, S.E.; Brooks, G.A.; Dahut, W.L.; Diasio, R.B. DPYD Testing: Time to Put Patient Safety First. J. Clin. Oncol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.D.; Hennebelle, I.; Delmas, C.; Lochon, I.; Tixidre, A.; Garnier, C.; Bonadona, A.; Penel, N.; Goncalves, E.C.; Delord, J.P.; et al. Genotyping of a family with a novel deleterious DPYD mutation supports the pretherapeutic screening of DPD deficiency with dihydrouracil/uracil ratio. Clin. Pharmacol. Ther. 2016, 99, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Nahid, N.A.; Apu, M.N.H.; Islam, R.; Shabnaz, S.; Chowdhury, S.M.; Ahmed, M.U.; Nahar, Z.; Islam, S.; Islam, M.S.; Hasnat, A. DPYD*2A and MTHFR C677T predict toxicity and efficacy, respectively, in patients on chemotherapy with 5-fluorouracil for colorectal cancer. Cancer Chemother. Pharmacol. 2018, 81, 119–129. [Google Scholar] [CrossRef]
- Clarke, W.A.; Chatelut, E.; Fotoohi, A.K.; Larson, R.A.; Martin, J.H.; Mathijssen, R.H.; Salamone, S.J. Therapeutic drug monitoring in oncology: International Association of Therapeutic Drug Monitoring and Clinical Toxicology consensus guidelines for imatinib therapy. Eur. J. Cancer 2021, 157, 428–440. [Google Scholar] [CrossRef]
- Beumer, J.H.; Chu, E.; Allegra, C.; Tanigawara, Y.; Milano, G.; Diasio, R.; Kim, T.W.; Mathijssen, R.H.; Zhang, L.; Arnold, D.; et al. Therapeutic Drug Monitoring in Oncology: International Association of Therapeutic Drug Monitoring and Clinical Toxicology Recommendations for 5-Fluorouracil Therapy. Clin. Pharmacol. Ther. 2019, 105, 598–613. [Google Scholar] [CrossRef]
- Rossi, T.; Bandini, E.; Balzi, W.; Fabbri, F.; Massa, I.; Maltoni, R. Obesity and Dose of Anti-cancer Therapy: Are We Sure to Be on the Right Track in the Precision Medicine Era? Front. Med. 2021, 8, 16–19. [Google Scholar] [CrossRef]
- Silvestris, N.; Argentiero, A.; Natalicchio, A.; D’Oronzo, S.; Beretta, G.; Acquati, S.; Adinolfi, V.; Di Bartolo, P.; Danesi, R.; Faggiano, A.; et al. Antineoplastic dosing in overweight and obese cancer patients: An Associazione Italiana Oncologia Medica (AIOM)/Associazione Medici Diabetologi (AMD)/Società Italiana Endocrinologia (SIE)/Società Italiana Farmacologia (SIF) multidisciplinary consensus pos. ESMO Open 2021, 6, 100153. [Google Scholar] [CrossRef]
- Hall, R.G.; Jean, G.W.; Sigler, M.; Shah, S. Dosing Considerations for Obese Patients Receiving Cancer Chemotherapeutic Agents. Ann. Pharmacother. 2013, 47, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Mindt, S.; Aida, S.; Merx, K.; Müller, A.; Gutting, T.; Hedtke, M.; Neumaier, M.; Hofheinz, R.-D. Therapeutic Drug Monitoring (TDM) of 5-fluorouracil (5-FU): New Preanalytic Aspects. Clin. Chem. Lab. Med. 2019, 57, 1012–1016. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | N = 50 (%) |
|---|---|
| Age. mean (SD) | 54.08 (12.75) |
| Sex Female Male | 30 (60) 20 (40) |
| BSA, Median (Q1, Q3) | 1.63 (1.49, 1.71) |
| BMI, Median (Q1, Q3) | 22.7 (19.8, 27.6) |
| HIV Status Positive Negative | 11 (22) 39 (78) |
| Tumour site N (%) Colorectal Breast Gastric and oesophagus Cervical Pancreas Others (Head & neck and liver) | 26 (52) 9 (18) 8 (16) 3 (6) 2 (4) 2(4) |
| Chemotherapy regimen CAPEOX CAPE 5-FU + Cisplatin CAPE + Cisplatin CAPE + GEM 5-FU + Others a | 29 (58) 12 (24) 3 (6) 1 (2) 2 (4) 3 (6) |
| AJCC Group Staging I II III IV | 2 (4) 11 (22) 14 (28) 23 (46) |
| Variant | RsID | Nucleotide Change | Single AA Change | DPD Activity | America a | Europe a | SAS a | Africa a | Zimbabwe b | This Cohort |
|---|---|---|---|---|---|---|---|---|---|---|
| *2A | rs3918290 | c.190511G>A | Not changed | No activity | 0.0010 | 0.0045 | 0.0034 | 0.0007 | 0.0019 | 0.0000 |
| *13 | rs55886062 | c.1679T>G | p.I560S | No activity | 0.0000 | 0.0010 | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| NA | rs67376798 | c.2846A>T | p.D949V | Decreased activity | 0.0030 | 0.0070 | 0.0010 | 0.0008 | 0.0029 | 0.0000 |
| Hap B3 | rs75017182 | c.1129-5923C>G | Not changed | Decreased activity | 0.0060 | 0.0239 | 0.0190 | 0.0008 | NA | 0.0000 |
| Type of population | NA | NA | NA | NA | General population | General population | General population | General population | General population | Focused Population |
| Population size | NA | NA | NA | NA | 694 | 1006 | 978 | 1322 | 522 | 150 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afolabi, B.L.; Mazhindu, T.; Zedias, C.; Borok, M.; Ndlovu, N.; Masimirembwa, C.; on behalf of Consortium for Genomics and Therapeutics in Africa (CGTA). Pharmacogenetics and Adverse Events in the Use of Fluoropyrimidine in a Cohort of Cancer Patients on Standard of Care Treatment in Zimbabwe. J. Pers. Med. 2023, 13, 588. https://doi.org/10.3390/jpm13040588
Afolabi BL, Mazhindu T, Zedias C, Borok M, Ndlovu N, Masimirembwa C, on behalf of Consortium for Genomics and Therapeutics in Africa (CGTA). Pharmacogenetics and Adverse Events in the Use of Fluoropyrimidine in a Cohort of Cancer Patients on Standard of Care Treatment in Zimbabwe. Journal of Personalized Medicine. 2023; 13(4):588. https://doi.org/10.3390/jpm13040588
Chicago/Turabian StyleAfolabi, Boluwatife Lawrence, Tinashe Mazhindu, Chikwambi Zedias, Margaret Borok, Ntokozo Ndlovu, Collen Masimirembwa, and on behalf of Consortium for Genomics and Therapeutics in Africa (CGTA). 2023. "Pharmacogenetics and Adverse Events in the Use of Fluoropyrimidine in a Cohort of Cancer Patients on Standard of Care Treatment in Zimbabwe" Journal of Personalized Medicine 13, no. 4: 588. https://doi.org/10.3390/jpm13040588