
The Rationale for Vitamin, Mineral, and Cofactor Treatment in the Precision Medical Care of Autism Spectrum Disorder

 , ,
, ,  ,
,  ,
,  ,
, 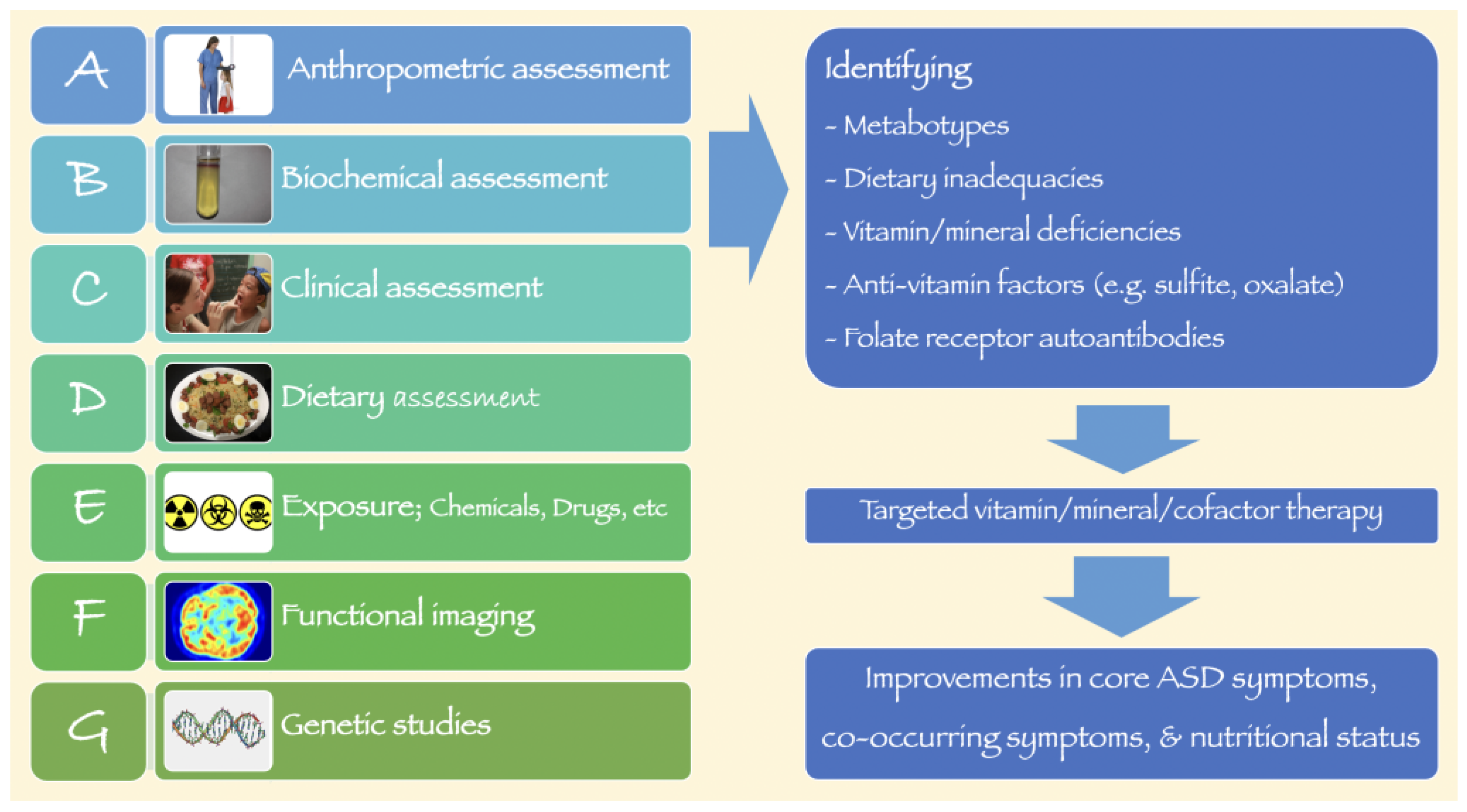{kind=link}
Abstract
:1. Introduction
2. Vitamins
2.1. Vitamin B1
2.2. Vitamin B2
2.3. Vitamin B3
2.4. Vitamin B5
2.5. Vitamin B6
2.6. Vitamin B7
2.7. Vitamin B9
2.8. Vitamin B12
2.9. Vitamin C
2.10. Vitamin E
2.11. Vitamin D
2.12. Vitamin A (Retinol)
3. Minerals
3.1. Zinc
3.2. Magnesium
3.3. Vitamin B6-Magnesium Combination
3.4. Molybdenum
3.5. Selenium
3.6. Copper
3.7. Iron
4. Other Cofactors
4.1. Coenzyme Q10
4.2. Alpha-Lipoic Acid
4.3. Tetrahydrobiopterin
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®); American Psychiatric Pub: Washington, DC, USA, 2013; Available online: https://www.psychiatry.org/psychiatrists/practice/dsm (accessed on 15 November 2022).
- Salari, N.; Rasoulpoor, S.; Rasoulpoor, S.; Shohaimi, S.; Jafarpour, S.; Abdoli, N.; Khaledi-Paveh, B.; Mohammadi, M. The global prevalence of autism spectrum disorder: A comprehensive systematic review and meta-analysis. Ital. J. Pediatr. 2022, 48, 112. [Google Scholar] [CrossRef] [PubMed]
- Devanand, S.; Manoli, M.D.; Matthew, W.; State, M.D. Autism Spectrum Disorder Genetics and the Search for Pathological Mechanisms. Am. J. Psychiatry 2021, 178, 30–38. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of Genetic and Environmental Factors with Autism in a 5-Country Cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef]
- Bhandari, R.; Paliwal, J.K.; Kuhad, A. Neuropsychopathology of Autism Spectrum Disorder: Complex Interplay of Genetic, Epigenetic, and Environmental Factors. Adv. Neurobiol. 2020, 24, 97–141. [Google Scholar] [CrossRef]
- Howes, O.D.; Rogdaki, M.; Findon, J.L.; Wichers, R.H.; Charman, T.; King, B.H.; Loth, E.; McAlonan, G.M.; McCracken, J.T.; Parr, J.R. Autism spectrum disorder: Consensus guidelines on assessment, treatment and research from the British Association for Psychopharmacology. J. Psychopharmacol. Oxf. Engl. 2018, 32, 3. [Google Scholar] [CrossRef]
- Meguid, N.; Anwar, M.; Zaki, S.; Kandeel, W.; Ahmed, N.; Tewfik, I. Dietary Patterns of Children with Autism Spectrum Disorder: A Study Based in Egypt. Open Access Maced. J. Med. Sci. 2015, 3, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Esteban-Figuerola, P.; Canals, J.; Fernández-Cao, J.C.; Arija Val, V. Differences in food consumption and nutritional intake between children with autism spectrum disorders and typically developing children: A meta-analysis. Autism 2019, 23, 1079–1095. [Google Scholar] [CrossRef]
- Chen, L.; Shi, X.J.; Liu, H.; Mao, X.; Gui, L.N.; Wang, H.; Cheng, Y. Oxidative stress marker aberrations in children with autism spectrum disorder: A systematic review and meta-analysis of 87 studies (N = 9109). Transl. Psychiatry 2021, 11, 15. [Google Scholar] [CrossRef]
- Saghazadeh, A.; Ahangari, N.; Hendi, K.; Saleh, F.; Rezaei, N. Status of essential elements in autism spectrum disorder: Systematic review and meta-analysis. Rev. Neurosci. 2017, 28, 783–809. [Google Scholar] [CrossRef]
- Zhu, J.; Guo, M.; Yang, T.; Lai, X.; Tang, T.; Chen, J.; Li, L.; Li, T. Nutritional Status and Symptoms in Preschool Children with Autism Spectrum Disorder: A Two-Center Comparative Study in Chongqing and Hainan Province, China. Front. Pediatr. 2020, 8, 469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hodgson, N.W.; Trivedi, M.S.; Abdolmaleky, H.M.; Fournier, M.; Cuenod, M.; Do, K.Q.; Deth, R.C. Decreased Brain Levels of Vitamin B12 in Aging, Autism and Schizophrenia. PLoS ONE 2016, 11, e0146797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossignol, D.A.; Frye, R.E. Cerebral Folate Deficiency, Folate Receptor Alpha Autoantibodies and Leucovorin (Folinic Acid) Treatment in Autism Spectrum Disorders: A Systematic Review and Meta-Analysis. J. Pers. Med. 2021, 11, 1141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Choi, M.J.; Ha, S.; Hwang, J.; Koyanagi, A.; Dragioti, E.; Radua, J.; Smith, L.; Jacob, L.; Salazar de Pablo, G.; et al. Association between autism spectrum disorder and inflammatory bowel disease: A systematic review and meta-analysis. Autism Res. 2022, 15, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.M.; Shenkin, A.; Schweinlin, A.; Amrein, K.; Augsburger, M.; Biesalski, H.K.; Bischoff, S.C.; Casaer, M.P.; Gundogan, K.; Lepp, H.L.; et al. ESPEN micronutrient guideline. Clin. Nutr. 2022, 41, 1357–1424. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Tessing, J.; Lee, B.K.; Lyall, K. Maternal Dietary Factors and the Risk of Autism Spectrum Disorders: A Systematic Review of Existing Evidence. Autism Res. 2020, 13, 1634–1658. [Google Scholar] [CrossRef]
- Karhu, E.; Zukerman, R.; Eshraghi, R.S.; Mittal, J.; Deth, R.C.; Castejon, A.M.; Trivedi, M.; Mittal, R.; Eshraghi, A.A. Nutritional interventions for autism spectrum disorder. Nutr. Rev. 2020, 78, 515–531. [Google Scholar] [CrossRef]
- Trudeau, M.S.; Madden, R.F.; Parnell, J.A.; Gibbard, W.B.; Shearer, J. Dietary and Supplement-Based Complementary and Alternative Medicine Use in Pediatric Autism Spectrum Disorder. Nutrients 2019, 11, 1783. [Google Scholar] [CrossRef] [Green Version]
- Stewart, P.A.; Hyman, S.L.; Schmidt, B.L.; Macklin, E.A.; Reynolds, A.; Johnson, C.R.; James, S.J.; Manning-Courtney, P. Dietary Supplementation in Children with Autism Spectrum Disorders: Common, Insufficient, and Excessive. J. Acad. Nutr. Diet. 2015, 115, 1237–1248. [Google Scholar] [CrossRef]
- Adams, J.B.; Audhya, T.; McDonough-Means, S.; Rubin, R.A.; Quig, D.; Geis, E.; Gehn, E.; Loresto, M.; Mitchell, J.; Atwood, S.; et al. Effect of a vitamin/mineral supplement on children and adults with autism. BMC Pediatr. 2011, 11, 111. [Google Scholar] [CrossRef]
- Legido, A.; Goldenthal, M.; Garvin, B.; Damle, S.; Corrigan, K.; Connell, J.; Thao, D.; Valencia, I.; Melvin, J.; Khurana, D.; et al. Effect of a Combination of Carnitine, Coenzyme Q10 and Alpha-Lipoic Acid (MitoCocktail) on Mitochondrial Function and Neurobehavioral Performance in Children with Autism Spectrum Disorder (P3.313). Neurology 2018, 90, P3.313. [Google Scholar]
- Bettendorff, L.; Wins, P. Biochemistry of Thiamine and Thiamine Phosphate Compounds. In Encyclopedia of Biological Chemistry III, 3rd ed.; Jez, J., Ed.; Elsevier: Oxford, UK, 2021; pp. 302–313. [Google Scholar]
- Mkrtchyan, G.; Aleshin, V.; Parkhomenko, Y.; Kaehne, T.; Di Salvo, M.L.; Parroni, A.; Contestabile, R.; Vovk, A.; Bettendorff, L.; Bunik, V. Molecular mechanisms of the non-coenzyme action of thiamin in brain: Biochemical, structural and pathway analysis. Sci. Rep. 2015, 5, 12583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleshin, V.A.; Mkrtchyan, G.V.; Bunik, V.I. Mechanisms of Non-coenzyme Action of Thiamine: Protein Targets and Medical Significance. Biochem. Mosc. 2019, 84, 829–850. [Google Scholar] [CrossRef]
- Wiley, K.D.; Gupta, M. Vitamin B1 Thiamine Deficiency. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Adams, J.B.; Audhya, T.; McDonough-Means, S.; Rubin, R.A.; Quig, D.; Geis, E.; Gehn, E.; Loresto, M.; Mitchell, J.; Atwood, S.; et al. Nutritional and metabolic status of children with autism vs. neurotypical children, and the association with autism severity. Nutr. Metab. Lond. 2011, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anwar, A.; Marini, M.; Abruzzo, P.M.; Bolotta, A.; Ghezzo, A.; Visconti, P.; Thornalley, P.J.; Rabbani, N. Quantitation of plasma thiamine, related metabolites and plasma protein oxidative damage markers in children with autism spectrum disorder and healthy controls. Free. Radic. Res. 2016, 50, S85–S90. [Google Scholar] [CrossRef] [Green Version]
- Yule, S.; Wanik, J.; Holm, E.M.; Bruder, M.B.; Shanley, E.; Sherman, C.Q.; Fitterman, M.; Lerner, J.; Marcello, M.; Parenchuck, N.; et al. Nutritional Deficiency Disease Secondary to ARFID Symptoms Associated with Autism and the Broad Autism Phenotype: A Qualitative Systematic Review of Case Reports and Case Series. J. Acad. Nutr. Diet. 2021, 121, 467–492. [Google Scholar] [CrossRef]
- Lonsdale, D. Thiamin. Adv. Food Nutr. Res. 2018, 83, 1–56. [Google Scholar] [CrossRef]
- Kahathuduwa, C.N.; West, B.D.; Blume, J.; Dharavath, N.; Moustaid-Moussa, N.; Mastergeorge, A. The risk of overweight and obesity in children with autism spectrum disorders: A systematic review and meta-analysis. Obes. Rev. 2019, 20, 1667–1679. [Google Scholar] [CrossRef]
- Sammels, O.; Karjalainen, L.; Dahlgren, J.; Wentz, E. Autism Spectrum Disorder and Obesity in Children: A Systematic Review and Meta-Analysis. Obes. Facts 2022, 15, 305–320. [Google Scholar] [CrossRef]
- Kerns, J.C.; Arundel, C.; Chawla, L.S. Thiamin deficiency in people with obesity. Adv. Nutr. 2015, 6, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Densupsoontorn, N.; Srisawat, C.; Chotipanang, K.; Junnu, S.; Kunnangja, S.; Wongarn, R.; Sriboonnark, W.; Tirapongporn, H.; Phuangphan, P. Prevalence of and factors associated with thiamin deficiency in obese Thai children. Asia Pac. J. Clin. Nutr. 2019, 28, 116–121. [Google Scholar] [CrossRef]
- Lonsdale, D. A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its derivatives. Evid.-Based Complement. Altern. Med. 2006, 3, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdale, D. The role of thiamin in high calorie malnutrition. Austin J. Nutr. Food Sci. 2015, 3, 1061. [Google Scholar]
- Indika, N.R.; Deutz, N.E.P.; Engelen, M.; Peiris, H.; Wijetunge, S.; Perera, R. Sulfur amino acid metabolism and related metabotypes of autism spectrum disorder: A review of biochemical evidence for a hypothesis. Biochimie 2021, 184, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Thompson, M.; Sullivan, N.; Child, G. Thiamine deficiency in dogs due to the feeding of sulphite preserved meat. Aust. Vet. J. 2005, 83, 412–417. [Google Scholar] [CrossRef]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef] [Green Version]
- Plantone, D.; Pardini, M.; Rinaldi, G. Riboflavin in Neurological Diseases: A Narrative Review. Clin. Drug Investig. 2021, 41, 513–527. [Google Scholar] [CrossRef]
- Tardy, A.L.; Pouteau, E.; Marquez, D.; Yilmaz, C.; Scholey, A. Vitamins and Minerals for Energy, Fatigue and Cognition: A Narrative Review of the Biochemical and Clinical Evidence. Nutrients 2020, 12, 228. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.; Wong, A. Nicotinamide: An Update and Review of Safety & Differences from Niacin. Ski. Ther. Lett. 2020, 25, 7–11. [Google Scholar]
- Zaenglein, A.; Martin, A.; Carlson, L.; Williams, K.E. Pellagra secondary to selective eating in a child with autism. Pediatr. Dermatol. 2020, 37, 698–700. [Google Scholar] [CrossRef]
- Yap, I.K.; Angley, M.; Veselkov, K.A.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Urinary metabolic phenotyping differentiates children with autism from their unaffected siblings and age-matched controls. J. Proteome Res. 2010, 9, 2996–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Ke, X.; Xiao, Z.; Zhang, Y.; Chen, Y.; Li, Y.; Wang, Z.; Lin, L.; Yao, P.; Lu, J. Untargeted Metabolomic Profiling Using UHPLC-QTOF/MS Reveals Metabolic Alterations Associated with Autism. Biomed. Res. Int. 2020, 2020, 6105608. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.J.; Li, D.; Ma, Q.; Gu, X.Y.; Guo, M.; Lun, Y.Z.; Sun, W.P.; Wang, X.Y.; Cao, Y.; Zhou, S.S. Excess nicotinamide increases plasma serotonin and histamine levels. Sheng Li Xue Bao 2013, 65, 33–38. [Google Scholar] [PubMed]
- Guo, B.Q.; Ding, S.B.; Li, H.B. Blood biomarker levels of methylation capacity in autism spectrum disorder: A systematic review and meta-analysis. Acta Psychiatr. Scand. 2020, 141, 492–509. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, L.Q.; He, Z.X.; He, X.X.; Wang, Y.J.; Jian, Y.L.; Wang, X.; Zhang, B.B.; Su, C.; Lu, J.; et al. Autism candidate gene DIP2A regulates spine morphogenesis via acetylation of cortactin. PLoS Biol. 2019, 17, e3000461. [Google Scholar] [CrossRef]
- Uchida, Y.; Ito, K.; Ohtsuki, S.; Kubo, Y.; Suzuki, T.; Terasaki, T. Major involvement of Na+-dependent multivitamin transporter (SLC5A6/SMVT) in uptake of biotin and pantothenic acid by human brain capillary endothelial cells. J. Neurochem. 2015, 134, 97–112. [Google Scholar] [CrossRef]
- Castro, K.; Faccioli, L.S.; Baronio, D.; Gottfried, C.; Perry, I.S.; Riesgo, R. Feeding behavior and dietary intake of male children and adolescents with autism spectrum disorder: A case-control study. Int. J. Dev. Neurosci. 2016, 53, 68–74. [Google Scholar] [CrossRef]
- Lindsay, R.L.; Eugene Arnold, L.; Aman, M.G.; Vitiello, B.; Posey, D.J.; McDougle, C.J.; Scahill, L.; Pachler, M.; McCracken, J.T.; Tierney, E.; et al. Dietary status and impact of risperidone on nutritional balance in children with autism: A pilot study. J. Intellect. Dev. Disabil. 2006, 31, 204–209. [Google Scholar] [CrossRef]
- Liang, Y.; Xiao, Z.; Ke, X.; Yao, P.; Chen, Y.; Lin, L.; Lu, J. Urinary Metabonomic Profiling Discriminates Between Children with Autism and Their Healthy Siblings. Med. Sci. Monit. 2020, 26, e926634. [Google Scholar] [CrossRef]
- Gevi, F.; Zolla, L.; Gabriele, S.; Persico, A.M. Urinary metabolomics of young Italian autistic children supports abnormal tryptophan and purine metabolism. Mol. Autism 2016, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Tsujiguchi, H.; Miyagi, S.; Nguyen, T.T.T.; Hara, A.; Ono, Y.; Kambayashi, Y.; Shimizu, Y.; Nakamura, H.; Suzuki, K.; Suzuki, F.; et al. Relationship between Autistic Traits and Nutrient Intake among Japanese Children and Adolescents. Nutrients 2020, 12, 2258. [Google Scholar] [CrossRef] [PubMed]
- Ueland, P.M.; Ulvik, A.; Rios-Avila, L.; Midttun, Ø.; Gregory, J.F. Direct and Functional Biomarkers of Vitamin B6 Status. Annu. Rev. Nutr. 2015, 35, 33–70. [Google Scholar] [CrossRef] [PubMed]
- Wondrak, G.T.; Jacobson, E.L. Vitamin B6: Beyond coenzyme functions. Subcell Biochem. 2012, 56, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Clayton, P.T. B6-responsive disorders: A model of vitamin dependency. J. Inherit. Metab. Dis. 2006, 29, 317–326. [Google Scholar] [CrossRef]
- Sato, K. Why is vitamin B6 effective in alleviating the symptoms of autism? Med. Hypotheses 2018, 115, 103–106. [Google Scholar] [CrossRef]
- Huang, Y.; Su, L.; Wu, J. Pyridoxine Supplementation Improves the Activity of Recombinant Glutamate Decarboxylase and the Enzymatic Production of Gama-Aminobutyric Acid. PLoS ONE 2016, 11, e0157466. [Google Scholar] [CrossRef] [Green Version]
- Brondino, N.; Fusar-Poli, L.; Panisi, C.; Damiani, S.; Barale, F.; Politi, P. Pharmacological Modulation of GABA Function in Autism Spectrum Disorders: A Systematic Review of Human Studies. J. Autism Dev. Disord. 2016, 46, 825–839. [Google Scholar] [CrossRef]
- Liu, P.; Torrens-Spence, M.P.; Ding, H.; Christensen, B.M.; Li, J. Mechanism of cysteine-dependent inactivation of aspartate/glutamate/cysteine sulfinic acid α-decarboxylases. Amino Acids 2013, 44, 391–404. [Google Scholar] [CrossRef]
- Han, Y.; Xi, Q.Q.; Dai, W.; Yang, S.H.; Gao, L.; Su, Y.Y.; Zhang, X. Abnormal transsulfuration metabolism and reduced antioxidant capacity in Chinese children with autism spectrum disorders. Int. J. Dev. Neurosci. 2015, 46, 27–32. [Google Scholar] [CrossRef]
- Yang, J.H.; Friederich, M.W.; Ellsworth, K.A.; Frederick, A.; Foreman, E.; Malicki, D.; Dimmock, D.; Lenberg, J.; Prasad, C.; Yu, A.C.; et al. Expanding the phenotypic and molecular spectrum of NFS1-related disorders that cause functional deficiencies in mitochondrial and cytosolic iron-sulfur cluster containing enzymes. Hum. Mutat. 2022, 43, 305–315. [Google Scholar] [CrossRef]
- Rontani, P.; Perche, O.; Greetham, L.; Jullien, N.; Gepner, B.; Féron, F.; Nivet, E.; Erard-Garcia, M. Impaired expression of the COSMOC/MOCOS gene unit in ASD patient stem cells. Mol. Psychiatry 2021, 26, 1606–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.; Shen, S.; Zhang, J.; Jing, P. Effects of Vitamin B6 Deficiency on the Composition and Functional Potential of T Cell Populations. J. Immunol. Res. 2017, 2017, 2197975. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Aggarwal, S.; Rashanravan, B.; Lee, T. Th1- and Th2-like cytokines in CD4+ and CD8+ T cells in autism. J. Neuroimmunol. 1998, 85, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Stach, K.; Stach, W.; Augoff, K. Vitamin B6 in Health and Disease. Nutrients 2021, 13, 3229. [Google Scholar] [CrossRef] [PubMed]
- Loohuis, L.M.; Albersen, M.; de Jong, S.; Wu, T.; Luykx, J.J.; Jans, J.J.M.; Verhoeven-Duif, N.M.; Ophoff, R.A. The Alkaline Phosphatase (ALPL) Locus Is Associated with B6 Vitamer Levels in CSF and Plasma. Genes 2018, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.B.; George, F.; Audhya, T. Abnormally high plasma levels of vitamin B6 in children with autism not taking supplements compared to controls not taking supplements. J. Altern. Complement. Med. 2006, 12, 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelban, V.; Wilson, M.P.; Warman Chardon, J.; Vandrovcova, J.; Zanetti, M.N.; Zamba-Papanicolaou, E.; Efthymiou, S.; Pope, S.; Conte, M.R.; Abis, G.; et al. PDXK mutations cause polyneuropathy responsive to pyridoxal 5′-phosphate supplementation. Ann. Neurol. 2019, 86, 225–240. [Google Scholar] [CrossRef] [Green Version]
- Register, T.C.; Wuthier, R.E. Effect of vanadate, a potent alkaline phosphatase inhibitor, on 45Ca and 32Pi uptake by matrix vesicle-enriched fractions from chicken epiphyseal cartilage. J. Biol. Chem. 1984, 259, 3511–3518. [Google Scholar] [CrossRef]
- Konstantynowicz, J.; Porowski, T.; Zoch-Zwierz, W.; Wasilewska, J.; Kadziela-Olech, H.; Kulak, W.; Owens, S.C.; Piotrowska-Jastrzebska, J.; Kaczmarski, M. A potential pathogenic role of oxalate in autism. Eur. J. Paediatr. Neurol. 2012, 16, 485–491. [Google Scholar] [CrossRef]
- Rothstein, A.; Cabantchik, Z.I.; Knauf, P. Mechanism of anion transport in red blood cells: Role of membrane proteins. Fed. Proc. 1976, 35, 3–10. [Google Scholar]
- Solomon, L.R. Considerations in the use of B6 vitamers in hematologic disorders: I. Red cell transport and metabolism of pyridoxal. Blood 1982, 59, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Remigante, A.; Spinelli, S.; Pusch, M.; Sarikas, A.; Morabito, R.; Marino, A.; Dossena, S. Role of SLC4 and SLC26 solute carriers during oxidative stress. Acta Physiol. Oxf. 2022, 235, e13796. [Google Scholar] [CrossRef]
- Talwar, D.; Catchpole, A.; Wadsworth, J.M.; Toole, B.J.; McMillan, D.C. The relationship between plasma albumin, alkaline phosphatase and pyridoxal phosphate concentrations in plasma and red cells: Implications for assessing vitamin B6 status. Clin. Nutr. 2020, 39, 2824–2831. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, S.; Sugaya, K.; Hokama, S.; Oshiro, Y.; Uchida, A.; Morozumi, M.; Ogawa, Y. Effect of vitamin B6 deficiency on glyoxylate metabolism in rats with or without glyoxylate overload. Biomed. Res. 2006, 27, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.B.; Holloway, C. Pilot study of a moderate dose multivitamin/mineral supplement for children with autistic spectrum disorder. J. Altern. Complement. Med. 2004, 10, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Vrolijk, M.F.; Opperhuizen, A.; Jansen, E.; Hageman, G.J.; Bast, A.; Haenen, G. The vitamin B6 paradox: Supplementation with high concentrations of pyridoxine leads to decreased vitamin B6 function. Toxicol. In Vitro 2017, 44, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Oppici, E.; Fargue, S.; Reid, E.S.; Mills, P.B.; Clayton, P.T.; Danpure, C.J.; Cellini, B. Pyridoxamine and pyridoxal are more effective than pyridoxine in rescuing folding-defective variants of human alanine:glyoxylate aminotransferase causing primary hyperoxaluria type I. Hum. Mol. Genet. 2015, 24, 5500–5511. [Google Scholar] [CrossRef] [Green Version]
- Van den Eynde, M.D.G.; Scheijen, J.; Stehouwer, C.D.A.; Miyata, T.; Schalkwijk, C.G. Quantification of the B6 vitamers in human plasma and urine in a study with pyridoxamine as an oral supplement; pyridoxamine as an alternative for pyridoxine. Clin. Nutr. 2021, 40, 4624–4632. [Google Scholar] [CrossRef]
- Hadtstein, F.; Vrolijk, M. Vitamin B-6-Induced Neuropathy: Exploring the Mechanisms of Pyridoxine Toxicity. Adv. Nutr. 2021, 12, 1911–1929. [Google Scholar] [CrossRef]
- Pannemans, D.L.; van den Berg, H.; Westerterp, K.R. The influence of protein intake on vitamin B-6 metabolism differs in young and elderly humans. J. Nutr. 1994, 124, 1207–1214. [Google Scholar] [CrossRef]
- Sharma, P.; Han, S.M.; Gillies, N.; Thorstensen, E.B.; Goy, M.; Barnett, M.P.G.; Roy, N.C.; Cameron-Smith, D.; Milan, A.M. Circulatory and Urinary B-Vitamin Responses to Multivitamin Supplement Ingestion Differ between Older and Younger Adults. Nutrients 2020, 12, 3529. [Google Scholar] [CrossRef] [PubMed]
- Jen, M.; Yan, A.C. Syndromes associated with nutritional deficiency and excess. Clin. Dermatol. 2010, 28, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Marin-Valencia, I.; Roe, C.R.; Pascual, J.M. Pyruvate carboxylase deficiency: Mechanisms, mimics and anaplerosis. Mol. Genet. Metab. 2010, 101, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.X.; Chen, Y.; Guo, H.R.; Chen, G.F. Systematic Review and Bioinformatic Analysis of microRNA Expression in Autism Spectrum Disorder Identifies Pathways Associated with Cancer, Metabolism, Cell Signaling, and Cell Adhesion. Front. Psychiatry 2021, 12, 630876. [Google Scholar] [CrossRef]
- Frye, R.E. Metabolic and mitochondrial disorders associated with epilepsy in children with autism spectrum disorder. Epilepsy Behav. 2015, 47, 147–157. [Google Scholar] [CrossRef] [Green Version]
- İnci, A.; Özaslan, A.; Okur, İ.; Biberoğlu, G.; Güney, E.; Ezgü, F.S.; Tümer, L.; İşeri, E. Autism: Screening of inborn errors of metabolism and unexpected results. Autism Res. 2021, 14, 887–896. [Google Scholar] [CrossRef]
- Yoldaş, T.; Gürbüz, B.B.; Akar, H.T.; Özmert, E.N.; Coşkun, T. Autism spectrum disorder in patients with inherited metabolic disorders-a large sample from a tertiary center. Turk. J. Pediatr. 2021, 63, 767–779. [Google Scholar] [CrossRef]
- Kiykim, E.; Zeybek, C.A.; Zubarioglu, T.; Cansever, S.; Yalcinkaya, C.; Soyucen, E.; Aydin, A. Inherited metabolic disorders in Turkish patients with autism spectrum disorders. Autism Res. 2016, 9, 217–223. [Google Scholar] [CrossRef]
- Zaffanello, M.; Zamboni, G.; Fontana, E.; Zoccante, L.; Tatò, L. A case of partial biotinidase deficiency associated with autism. Child Neuropsychol. 2003, 9, 184–188. [Google Scholar] [CrossRef]
- Benke, P.J.; Duchowny, M.; McKnight, D. Biotin and Acetazolamide for Treatment of an Unusual Child with Autism Plus Lack of Nail and Hair Growth. Pediatr. Neurol. 2018, 79, 61–64. [Google Scholar] [CrossRef]
- Colamaria, V.; Burlina, A.B.; Gaburro, D.; Pajno-Ferrara, F.; Saudubray, J.M.; Merino, R.G.; Dalla Bernardina, B. Biotin-responsive infantile encephalopathy: EEG-polygraphic study of a case. Epilepsia 1989, 30, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Al-Owain, M.; Kaya, N.; Al-Shamrani, H.; Al-Bakheet, A.; Qari, A.; Al-Muaigl, S.; Ghaziuddin, M. Autism spectrum disorder in a child with propionic acidemia. JIMD Rep. 2013, 7, 63–66. [Google Scholar] [CrossRef] [Green Version]
- de la Bâtie, C.D.; Barbier, V.; Roda, C.; Brassier, A.; Arnoux, J.B.; Valayannopoulos, V.; Guemann, A.S.; Pontoizeau, C.; Gobin, S.; Habarou, F.; et al. Autism spectrum disorders in propionic acidemia patients. J. Inherit. Metab. Dis. 2018, 41, 623–629. [Google Scholar] [CrossRef]
- Cakar, N.E.; Yilmazbas, P. Cases of inborn errors of metabolism diagnosed in children with autism. Ideggyogy Sz 2021, 74, 67–72. [Google Scholar] [CrossRef]
- Lietzan, A.D.; St. Maurice, M. Insights into the carboxyltransferase reaction of pyruvate carboxylase from the structures of bound product and intermediate analogs. Biochem. Biophys. Res. Commun. 2013, 441, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Stathopoulos, S.; Gaujoux, R.; Lindeque, Z.; Mahony, C.; Van Der Colff, R.; Van Der Westhuizen, F.; O’Ryan, C. DNA Methylation Associated with Mitochondrial Dysfunction in a South African Autism Spectrum Disorder Cohort. Autism Res. 2020, 13, 1079–1093. [Google Scholar] [CrossRef]
- Sahin, K.; Orhan, C.; Karatoprak, S.; Tuzcu, M.; Deeh, P.B.D.; Ozercan, I.H.; Sahin, N.; Bozoglan, M.Y.; Sylla, S.; Ojalvo, S.P.; et al. Therapeutic Effects of a Novel Form of Biotin on Propionic Acid-Induced Autistic Features in Rats. Nutrients 2022, 14, 1280. [Google Scholar] [CrossRef]
- Schänzer, A.; Döring, B.; Ondrouschek, M.; Goos, S.; Garvalov, B.K.; Geyer, J.; Acker, T.; Neubauer, B.; Hahn, A. Stress-induced upregulation of SLC19A3 is impaired in biotin-thiamine-responsive basal ganglia disease. Brain Pathol. 2014, 24, 270–279. [Google Scholar] [CrossRef]
- Vlasova, T.I.; Stratton, S.L.; Wells, A.M.; Mock, N.I.; Mock, D.M. Biotin deficiency reduces expression of SLC19A3, a potential biotin transporter, in leukocytes from human blood. J. Nutr. 2005, 135, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Saleem, F.; Soos, M.P. Biotin Deficiency. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Frye, R.E.; Rossignol, D.A. Treatments for biomedical abnormalities associated with autism spectrum disorder. Front. Pediatr. 2014, 2, 66. [Google Scholar] [CrossRef] [Green Version]
- Frustaci, A.; Neri, M.; Cesario, A.; Adams, J.B.; Domenici, E.; Dalla Bernardina, B.; Bonassi, S. Oxidative stress-related biomarkers in autism: Systematic review and meta-analyses. Free. Radic. Biol. Med. 2012, 52, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.T.; Rothenberg, S.P.; Sequeira, J.M.; Opladen, T.; Blau, N.; Quadros, E.V.; Selhub, J. Autoantibodies to folate receptors in the cerebral folate deficiency syndrome. N. Engl. J. Med. 2005, 352, 1985–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, R.E.; Slattery, J.C.; Quadros, E.V. Folate metabolism abnormalities in autism: Potential biomarkers. Biomark. Med. 2017, 11, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; Rossignol, D.A.; Scahill, L.; McDougle, C.J.; Huberman, H.; Quadros, E.V. Treatment of Folate Metabolism Abnormalities in Autism Spectrum Disorder. Semin. Pediatr. Neurol. 2020, 35, 100835. [Google Scholar] [CrossRef] [PubMed]
- Vashi, P.; Edwin, P.; Popiel, B.; Lammersfeld, C.; Gupta, D. Methylmalonic Acid and Homocysteine as Indicators of Vitamin B-12 Deficiency in Cancer. PLoS ONE 2016, 11, e0147843. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Jernigan, S.; Cleves, M.A.; Halsted, C.H.; Wong, D.H.; Cutler, P.; Bock, K.; Boris, M.; Bradstreet, J.J.; et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141B, 947–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nashabat, M.; Maegawa, G.; Nissen, P.H.; Nexo, E.; Al-Shamrani, H.; Al-Owain, M.; Alfadhel, M. Long-term Outcome of 4 Patients with Transcobalamin Deficiency Caused by 2 Novel TCN2 Mutations. J. Pediatr. Hematol. Oncol. 2017, 39, e430–e436. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. The Effectiveness of Cobalamin (B12) Treatment for Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. J. Pers. Med. 2021, 11, 784. [Google Scholar] [CrossRef]
- Paul, C.; Brady, D.M. Comparative Bioavailability and Utilization of Particular Forms of B12 Supplements with Potential to Mitigate B12-related Genetic Polymorphisms. Integr. Med. Encinitas 2017, 16, 42–49. [Google Scholar]
- Wang, Y.; Liu, X.J.; Robitaille, L.; Eintracht, S.; MacNamara, E.; Hoffer, L.J. Effects of vitamin C and vitamin D administration on mood and distress in acutely hospitalized patients. Am. J. Clin. Nutr. 2013, 98, 705–711. [Google Scholar] [CrossRef] [Green Version]
- Ma, N.S.; Thompson, C.; Weston, S. Brief Report: Scurvy as a Manifestation of Food Selectivity in Children with Autism. J. Autism Dev. Disord. 2016, 46, 1464–1470. [Google Scholar] [CrossRef]
- Andrews, S.L.; Iyer, S.; Rodda, C.; Fitzgerald, J. Scurvy: A rare cause for limp in a child with autism spectrum disorder. J. Paediatr. Child Health 2018, 54, 1375–1377. [Google Scholar] [CrossRef]
- Golriz, F.; Donnelly, L.F.; Devaraj, S.; Krishnamurthy, R. Modern American scurvy—experience with vitamin C deficiency at a large children’s hospital. Pediatr. Radiol. 2017, 47, 214–220. [Google Scholar] [CrossRef]
- Dolske, M.C.; Spollen, J.; McKay, S.; Lancashire, E.; Tolbert, L. A preliminary trial of ascorbic acid as supplemental therapy for autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 1993, 17, 765–774. [Google Scholar] [CrossRef]
- Masci, D.; Rubino, C.; Basile, M.; Indolfi, G.; Trapani, S. When the limp has a dietary cause: A retrospective study on scurvy in a tertiary Italian pediatric hospital. Front. Pediatr. 2022, 10, 981908. [Google Scholar] [CrossRef]
- Sharp, W.G.; Berry, R.C.; Burrell, L.; Scahill, L.; McElhanon, B.O. Scurvy as a Sequela of Avoidant-Restrictive Food Intake Disorder in Autism: A Systematic Review. J. Dev. Behav. Pediatr. 2020, 41, 397–405. [Google Scholar] [CrossRef]
- Pangrazzi, L.; Balasco, L.; Bozzi, Y. Natural Antioxidants: A Novel Therapeutic Approach to Autism Spectrum Disorders? Antioxidants 2020, 9, 1186. [Google Scholar] [CrossRef]
- Al-Gadani, Y.; El-Ansary, A.; Attas, O.; Al-Ayadhi, L. Metabolic biomarkers related to oxidative stress and antioxidant status in Saudi autistic children. Clin. Biochem. 2009, 42, 1032–1040. [Google Scholar] [CrossRef]
- El-Ansary, A.; Bjørklund, G.; Chirumbolo, S.; Alnakhli, O.M. Predictive value of selected biomarkers related to metabolism and oxidative stress in children with autism spectrum disorder. Metab. Brain Dis. 2017, 32, 1209–1221. [Google Scholar] [CrossRef]
- Alabdali, A.; Al-Ayadhi, L.; El-Ansary, A. A key role for an impaired detoxification mechanism in the etiology and severity of autism spectrum disorders. Behav. Brain Funct. 2014, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Hassan, W.M.; Al-Ayadhi, L.; Bjørklund, G.; Alabdali, A.; Chirumbolo, S.; El-Ansary, A. The Use of Multi-parametric Biomarker Profiles May Increase the Accuracy of ASD Prediction. J. Mol. Neurosci. 2018, 66, 85–101. [Google Scholar] [CrossRef]
- Alkhalidy, H.; Abushaikha, A.; Alnaser, K.; Obeidat, M.D.; Al-Shami, I. Nutritional Status of Pre-school Children and Determinant Factors of Autism: A Case-Control Study. Front. Nutr. 2021, 8, 627011. [Google Scholar] [CrossRef]
- Morris, C.R.; Agin, M.C. Syndrome of allergy, apraxia, and malabsorption: Characterization of a neurodevelopmental phenotype that responds to omega 3 and vitamin E supplementation. Altern. Ther. Health Med. 2009, 15, 34–43. [Google Scholar]
- Gáll, Z.; Székely, O. Role of Vitamin D in Cognitive Dysfunction: New Molecular Concepts and Discrepancies between Animal and Human Findings. Nutrients 2021, 13, 3672. [Google Scholar] [CrossRef]
- Gallardo-Carrasco, M.C.; Jiménez-Barbero, J.A.; Bravo-Pastor, M.D.M.; Martin-Castillo, D.; Sánchez-Muñoz, M. Serum Vitamin D, Folate and Fatty Acid Levels in Children with Autism Spectrum Disorders: A Systematic Review and Meta-Analysis. J. Autism Dev. Disord. 2022, 52, 4708–4721. [Google Scholar] [CrossRef]
- Wang, T.; Shan, L.; Du, L.; Feng, J.; Xu, Z.; Staal, W.G.; Jia, F. Serum concentration of 25-hydroxyvitamin D in autism spectrum disorder: A systematic review and meta-analysis. Eur. Child Adolesc. Psychiatry 2016, 25, 341–350. [Google Scholar] [CrossRef]
- Srinivasan, S.; O’Rourke, J.; Bersche Golas, S.; Neumeyer, A.; Misra, M. Calcium and Vitamin D Supplement Prescribing Practices among Providers Caring for Children with Autism Spectrum Disorders: Are We Addressing Bone Health? Autism Res. Treat. 2016, 2016, 6763205. [Google Scholar] [CrossRef] [Green Version]
- Safahani, M.; Aligholi, H.; Asadi-Pooya, A.A. Management of antiepileptic drug–induced nutrition-related adverse effects. Neurol. Sci. 2020, 41, 3491–3502. [Google Scholar] [CrossRef]
- García-Serna, A.M.; Morales, E. Neurodevelopmental effects of prenatal vitamin D in humans: Systematic review and meta-analysis. Mol. Psychiatry 2020, 25, 2468–2481. [Google Scholar] [CrossRef]
- Uçar, N.; Grant, W.B.; Peraita-Costa, I.; Morales Suárez-Varela, M. How 25(OH)D Levels during Pregnancy Affect Prevalence of Autism in Children: Systematic Review. Nutrients 2020, 12, 2311. [Google Scholar] [CrossRef]
- Li, B.; Xu, Y.; Zhang, X.; Zhang, L.; Wu, Y.; Wang, X.; Zhu, C. The effect of vitamin D supplementation in treatment of children with autism spectrum disorder: A systematic review and meta-analysis of randomized controlled trials. Nutr. Neurosci. 2022, 25, 835–845. [Google Scholar] [CrossRef]
- Feng, J.Y.; Li, H.H.; Wang, B.; Shan, L.; Jia, F.Y. Successive clinical application of vitamin D and bumetanide in children with autism spectrum disorder: A case report. Med. Baltim. 2020, 99, e18661. [Google Scholar] [CrossRef]
- Jia, F.; Wang, B.; Shan, L.; Xu, Z.; Staal, W.G.; Du, L. Core symptoms of autism improved after vitamin D supplementation. Pediatrics 2015, 135, e196–e198. [Google Scholar] [CrossRef] [Green Version]
- Stalnaker, L.D.; Prasher, P.; Flesher, S. Rickets treatment improves more than bone health in toddler with autism spectrum disorder: A brief report. SAGE Open Med. Case Rep. 2019, 7, 2050313X19870026. [Google Scholar] [CrossRef] [Green Version]
- Jia, F.; Shan, L.; Wang, B.; Li, H.; Feng, J.; Xu, Z.; Saad, K. Fluctuations in clinical symptoms with changes in serum 25(OH) vitamin D levels in autistic children: Three cases report. Nutr. Neurosci. 2019, 22, 863–866. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, T. Relationship between serum folic acid and 25(OH)D levels and intelligence and core symptoms in children with autism. Wei Sheng Yan Jiu 2021, 50, 957–961. [Google Scholar] [CrossRef]
- Godar, D.E.; Merrill, S.J. Untangling the most probable role for vitamin D(3) in autism. Dermato-Endocrinology 2017, 9, e1387702. [Google Scholar] [CrossRef] [Green Version]
- Patrick, R.P.; Ames, B.N. Vitamin D hormone regulates serotonin synthesis. Part 1: Relevance for autism. Faseb J. 2014, 28, 2398–2413. [Google Scholar] [CrossRef] [Green Version]
- Trifonova, E.A.; Klimenko, A.I.; Mustafin, Z.S.; Lashin, S.A.; Kochetov, A.V. The mTOR Signaling Pathway Activity and Vitamin D Availability Control the Expression of Most Autism Predisposition Genes. Int. J. Mol. Sci. 2019, 20, 6332. [Google Scholar] [CrossRef] [Green Version]
- Cannell, J.J.; Grant, W.B. What is the role of vitamin D in autism? Dermato-Endocrinology 2013, 5, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Spiller, H.A.; Good, T.F.; Spiller, N.E.; Aleguas, A. Vitamin D exposures reported to US poison centers 2000–2014: Temporal trends and outcomes. Hum. Exp. Toxicol. 2016, 35, 457–461. [Google Scholar] [CrossRef]
- Kittana, M.; Ahmadani, A.; Stojanovska, L.; Attlee, A. The Role of Vitamin D Supplementation in Children with Autism Spectrum Disorder: A Narrative Review. Nutrients 2021, 14, 26. [Google Scholar] [CrossRef]
- Kerley, C.P.; Elnazir, B.; Greally, P.; Coghlan, D. Blunted serum 25(OH)D response to vitamin D(3) supplementation in children with autism. Nutr. Neurosci. 2020, 23, 537–542. [Google Scholar] [CrossRef]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A metabolism: An update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Zhu, J.; Yang, T.; Lai, X.; Liu, X.; Liu, J.; Chen, J.; Li, T. Vitamin A improves the symptoms of autism spectrum disorders and decreases 5-hydroxytryptamine (5-HT): A pilot study. Brain Res. Bull. 2018, 137, 35–40. [Google Scholar] [CrossRef]
- Hou, N.; Ren, L.; Gong, M.; Bi, Y.; Gu, Y.; Dong, Z.; Liu, Y.; Chen, J.; Li, T. Vitamin A deficiency impairs spatial learning and memory: The mechanism of abnormal CBP-dependent histone acetylation regulated by retinoic acid receptor alpha. Mol. Neurobiol. 2015, 51, 633–647. [Google Scholar] [CrossRef]
- Zhang, M.; Jiao, J.; Hu, X.; Yang, P.; Huang, Y.; Situ, M.; Guo, K.; Cai, J.; Huang, Y. Exploring the spatial working memory and visual perception in children with autism spectrum disorder and general population with high autism-like traits. PLoS ONE 2020, 15, e0235552. [Google Scholar] [CrossRef]
- Jiang, W.; Yu, Q.; Gong, M.; Chen, L.; Wen, E.Y.; Bi, Y.; Zhang, Y.; Shi, Y.; Qu, P.; Liu, Y.X.; et al. Vitamin A deficiency impairs postnatal cognitive function via inhibition of neuronal calcium excitability in hippocampus. J. Neurochem. 2012, 121, 932–943. [Google Scholar] [CrossRef]
- Golini, R.S.; Delgado, S.M.; Navigatore Fonzo, L.S.; Ponce, I.T.; Lacoste, M.G.; Anzulovich, A.C. Daily patterns of clock and cognition-related factors are modified in the hippocampus of vitamin A-deficient rats. Hippocampus 2012, 22, 1720–1732. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Liu, J.; Xiong, X.; Yang, T.; Hou, N.; Liang, X.; Chen, J.; Cheng, Q.; Li, T. Correlation between Nutrition and Symptoms: Nutritional Survey of Children with Autism Spectrum Disorder in Chongqing, China. Nutrients 2016, 8, 294. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Zhou, Y.; Sun, C.; Wang, J.; Wu, L. A preliminary study on nutritional status and intake in Chinese children with autism. Eur. J. Pediatr. 2010, 169, 1201–1206. [Google Scholar] [CrossRef]
- Bandini, L.G.; Anderson, S.E.; Curtin, C.; Cermak, S.; Evans, E.W.; Scampini, R.; Maslin, M.; Must, A. Food selectivity in children with autism spectrum disorders and typically developing children. J. Pediatr. 2010, 157, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Hyman, S.L.; Stewart, P.A.; Schmidt, B.; Cain, U.; Lemcke, N.; Foley, J.T.; Peck, R.; Clemons, T.; Reynolds, A.; Johnson, C.; et al. Nutrient intake from food in children with autism. Pediatrics 2012, 130, S145–S153. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Xia, W.; Zhao, Y.; Li, N.; Zhao, D.; Wu, L. Nutritional status survey of children with autism and typically developing children aged 4–6 years in Heilongjiang Province, China. J. Nutr. Sci. 2013, 2, e16. [Google Scholar] [CrossRef] [Green Version]
- Tekes, K.; Gyenge, M.; Hantos, M.; Csaba, G. Transgenerational hormonal imprinting caused by vitamin A and vitamin D treatment of newborn rats. Alterations in the biogenic amine contents of the adult brain. Brain Dev. 2009, 31, 666–670. [Google Scholar] [CrossRef]
- Hough, L.H.; Segal, S. Effects of developmental hyperserotonemia on the morphology of rat dentate nuclear neurons. Neuroscience 2016, 322, 178–194. [Google Scholar] [CrossRef]
- World Health Organization. Technical Note: Quality and Regulatory Considerations for the Use of Vitamin A Supplements in Public Health Programmes for Infants and Children Aged 6–59 Months; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Ziegler, E.; Filer, L. Present Knowledge in Nutrition; International Life Sciences Institute—Nutrition Foundation: Washington, DC, USA, 1996. [Google Scholar]
- Penniston, K.L.; Tanumihardjo, S.A. The acute and chronic toxic effects of vitamin A. Am. J. Clin. Nutr. 2006, 83, 191–201. [Google Scholar] [CrossRef] [Green Version]
- King, J.C.; Brown, K.H.; Gibson, R.S.; Krebs, N.F.; Lowe, N.M.; Siekmann, J.H.; Raiten, D.J. Biomarkers of Nutrition for Development (BOND)-Zinc Review. J. Nutr. 2015, 146, 858S–885S. [Google Scholar] [CrossRef] [Green Version]
- Bagherani, N.; Smoller, B.R. An overview of zinc and its importance in dermatology- Part I: Importance and function of zinc in human beings. Glob. Dermatol. 2016, 3, 330–336. [Google Scholar] [CrossRef]
- Sayehmiri, F.; Babaknejad, N.; Bahrami, S.; Sayehmiri, K.; Darabi, M.; Rezaei-Tavirani, M. Zn/Cu Levels in the Field of Autism Disorders: A Systematic Review and Meta-analysis. Iran. J. Child Neurol. 2015, 9, 1–9. [Google Scholar]
- Zhang, J.; Li, X.; Shen, L.; Khan, N.U.; Zhang, X.; Chen, L.; Zhao, H.; Luo, P. Trace elements in children with autism spectrum disorder: A meta-analysis based on case-control studies. J. Trace Elem. Med. Biol. 2021, 67, 126782. [Google Scholar] [CrossRef]
- Grabrucker, A.M. Zinc in the developing brain. In Nutrition and the Developing Brain; Moran, V.H., Lowe, N., Eds.; CRC Press: Boca Raton, FL, USA, 2016; pp. 143–168. [Google Scholar]
- Maywald, M.; Wessels, I.; Rink, L. Zinc Signals and Immunity. Int. J. Mol. Sci. 2017, 18, 2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltaci, A.K.; Mogulkoc, R.; Baltaci, S.B. Review: The role of zinc in the endocrine system. Pak. J. Pharm. Sci. 2019, 32, 231–239. [Google Scholar]
- Sauer, A.K.; Malijauskaite, S.; Meleady, P.; Boeckers, T.M.; McGourty, K.; Grabrucker, A.M. Zinc is a key regulator of gastrointestinal development, microbiota composition and inflammation with relevance for autism spectrum disorders. Cell Mol. Life Sci. 2021, 79, 46. [Google Scholar] [CrossRef] [PubMed]
- Sauer, A.K.; Grabrucker, A.M. Zinc Deficiency During Pregnancy Leads to Altered Microbiome and Elevated Inflammatory Markers in Mice. Front. Neurosci. 2019, 13, 1295. [Google Scholar] [CrossRef]
- Grabrucker, A. The specific role of zinc in autism spectrum disorders. In Biometals in Autism Spectrum Disorders; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Grabrucker, S.; Jannetti, L.; Eckert, M.; Gaub, S.; Chhabra, R.; Pfaender, S.; Mangus, K.; Reddy, P.P.; Rankovic, V.; Schmeisser, M.J.; et al. Zinc deficiency dysregulates the synaptic ProSAP/Shank scaffold and might contribute to autism spectrum disorders. Brain 2014, 137, 137–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabrucker, S.; Boeckers, T.M.; Grabrucker, A.M. Gender Dependent Evaluation of Autism like Behavior in Mice Exposed to Prenatal Zinc Deficiency. Front. Behav. Neurosci. 2016, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Grabrucker, S.; Haderspeck, J.C.; Sauer, A.K.; Kittelberger, N.; Asoglu, H.; Abaei, A.; Rasche, V.; Schön, M.; Boeckers, T.M.; Grabrucker, A.M. Brain Lateralization in Mice Is Associated with Zinc Signaling and Altered in Prenatal Zinc Deficient Mice That Display Features of Autism Spectrum Disorder. Front. Mol. Neurosci. 2017, 10, 450. [Google Scholar] [CrossRef]
- Sauer, A.K.; Dooley, L.; Vaughan, A.; Grabrucker, A.M. Altered gut–brain signaling in autism spectrum disorders—From biomarkers to possible intervention strategies. In Neural Engineering Techniques for Autism Spectrum Disorder; Academic Press: Cambridge, MA, USA, 2021; pp. 127–149. [Google Scholar]
- Hagmeyer, S.; Sauer, A.K.; Grabrucker, A.M. Prospects of Zinc Supplementation in Autism Spectrum Disorders and Shankopathies Such as Phelan McDermid Syndrome. Front. Synaptic Neurosci. 2018, 10, 11. [Google Scholar] [CrossRef]
- Fourie, C.; Vyas, Y.; Lee, K.; Jung, Y.; Garner, C.C.; Montgomery, J.M. Dietary Zinc Supplementation Prevents Autism Related Behaviors and Striatal Synaptic Dysfunction in Shank3 Exon 13-16 Mutant Mice. Front. Cell Neurosci. 2018, 12, 374. [Google Scholar] [CrossRef] [Green Version]
- Vyas, Y.; Lee, K.; Jung, Y.; Montgomery, J.M. Influence of maternal zinc supplementation on the development of autism-associated behavioural and synaptic deficits in offspring Shank3-knockout mice. Mol. Brain 2020, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Lee, H.; Huang, T.N.; Chung, C.; Shin, W.; Kim, K.; Koh, J.Y.; Hsueh, Y.P.; Kim, E. Trans-synaptic zinc mobilization improves social interaction in two mouse models of autism through NMDAR activation. Nat. Commun. 2015, 6, 7168. [Google Scholar] [CrossRef] [Green Version]
- Cezar, L.C.; Kirsten, T.B.; da Fonseca, C.C.N.; de Lima, A.P.N.; Bernardi, M.M.; Felicio, L.F. Zinc as a therapy in a rat model of autism prenatally induced by valproic acid. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 84, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Jung, Y.; Vyas, Y.; Skelton, I.; Abraham, W.C.; Hsueh, Y.P.; Montgomery, J.M. Dietary zinc supplementation rescues fear-based learning and synaptic function in the Tbr1(+/-) mouse model of autism spectrum disorders. Mol. Autism 2022, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Schoendorfer, N.; Davies, P. Micronutrient interrelationships: Synergism and antagonism. In Micronutrients, 1st ed.; Betancourt, A.I., Gaitan, H.F., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2012; pp. 159–177. [Google Scholar]
- Potocnik, F.C.; van Rensburg, S.J.; Hon, D.; Emsley, R.A.; Moodie, I.M.; Erasmus, R.T. Oral zinc augmentation with vitamins A and D increases plasma zinc concentration: Implications for burden of disease. Metab. Brain Dis. 2006, 21, 139–147. [Google Scholar] [CrossRef]
- de Baaij, J.H.; Hoenderop, J.G.; Bindels, R.J. Magnesium in man: Implications for health and disease. Physiol. Rev. 2015, 95, 1–46. [Google Scholar] [CrossRef]
- Botturi, A.; Ciappolino, V.; Delvecchio, G.; Boscutti, A.; Viscardi, B.; Brambilla, P. The Role and the Effect of Magnesium in Mental Disorders: A Systematic Review. Nutrients 2020, 12, 1661. [Google Scholar] [CrossRef]
- Kotb, M.; Kredich, N.M. S-Adenosylmethionine synthetase from human lymphocytes. Purification and characterization. J. Biol. Chem. 1985, 260, 3923–3930. [Google Scholar] [CrossRef]
- Brudnak, M.; Buchholz, I.; Hoener, S.; Newman, L.; Pangborn, J. Guide to Intestinal Health in Autism Spectrum Disorder; Kirkman Laboratories: Lake Oswego, OR, USA, 2001. [Google Scholar]
- Waring, R.H.; Klovrza, L.V. Sulphur Metabolism in Autism. J. Nutr. Environ. Med. 2000, 10, 25–32. [Google Scholar] [CrossRef]
- Adams, J.B.; Audhya, T.; Geis, E.; Gehn, E.; Fimbres, V.; Pollard, E.L.; Mitchell, J.; Ingram, J.; Hellmers, R.; Laake, D.; et al. Comprehensive Nutritional and Dietary Intervention for Autism Spectrum Disorder—A Randomized, Controlled 12-Month Trial. Nutrients 2018, 10, 369. [Google Scholar] [CrossRef] [Green Version]
- Peake, R.W.; Godber, I.M.; Maguire, D. The effect of magnesium administration on erythrocyte transketolase activity in alcoholic patients treated with thiamine. Scott. Med. J. 2013, 58, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.E.; Schwartz, U.D.; Lubran, M.M. Effect of vitamin B-6 on plasma and red blood cell magnesium levels in premenopausal women. Ann. Clin. Lab. Sci. 1981, 11, 333–336. [Google Scholar]
- Boylan, L.M.; Spallholz, J.E. In vitro evidence for a relationship between magnesium and vitamin B-6. Magnes. Res. 1990, 3, 79–85. [Google Scholar] [PubMed]
- Bunik, V.; Aleshin, V.; Nogues, I.; Kähne, T.; Parroni, A.; Contestabile, R.; Salvo, M.L.; Graf, A.; Tramonti, A. Thiamine-dependent regulation of mammalian brain pyridoxal kinase in vitro and in vivo. J. Neurochem. 2022, 161, 20–39. [Google Scholar] [CrossRef]
- Rimland, B.; Callaway, E.; Dreyfus, P. The effect of high doses of vitamin B6 on autistic children: A double-blind crossover study. Am. J. Psychiatry 1978, 135, 472–475. [Google Scholar] [CrossRef]
- Nye, C.; Brice, A. Combined vitamin B6-magnesium treatment in autism spectrum disorder. Cochrane Database Syst. Rev. 2005, 2005, CD003497. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.; Rahman, M.S.; Akhter, S.; Momen, A.B.I.; Raihan, S.G. Vitamin B6 and magnesium on neurobehavioral status of autism spectrum disorder: A randomized, double-blind, placebo controlled study. Bangladesh J. Med. 2021, 32, 12–18. [Google Scholar] [CrossRef]
- Mousain-Bosc, M.; Roche, M.; Polge, A.; Pradal-Prat, D.; Rapin, J.; Bali, J.P. Improvement of neurobehavioral disorders in children supplemented with magnesium-vitamin B6. II. Pervasive developmental disorder-autism. Magnes. Res. 2006, 19, 53–62. [Google Scholar]
- Huijmans, J.G.M.; Schot, R.; de Klerk, J.B.C.; Williams, M.; de Coo, R.F.M.; Duran, M.; Verheijen, F.W.; van Slegtenhorst, M.; Mancini, G.M.S. Molybdenum cofactor deficiency: Identification of a patient with homozygote mutation in the MOCS3 gene. Am. J. Med. Genet. A 2017, 173, 1601–1606. [Google Scholar] [CrossRef]
- Lionel, A.C.; Vaags, A.K.; Sato, D.; Gazzellone, M.J.; Mitchell, E.B.; Chen, H.Y.; Costain, G.; Walker, S.; Egger, G.; Thiruvahindrapuram, B.; et al. Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum. Mol. Genet. 2013, 22, 2055–2066. [Google Scholar] [CrossRef] [Green Version]
- Raymond, L.J.; Deth, R.C.; Ralston, N.V. Potential Role of Selenoenzymes and Antioxidant Metabolism in relation to Autism Etiology and Pathology. Autism Res. Treat. 2014, 2014, 164938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Zhao, G.; Liu, S.; Zhang, Q.; Wang, P.; Cao, Y.; Wu, L. Supplementation with selenium attenuates autism-like behaviors and improves oxidative stress, inflammation and related gene expression in an autism disease model. J. Nutr. Biochem. 2022, 107, 109034. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Luan, Y.; Wang, H.; Zhang, P.; Liu, S.; Wang, P.; Cao, Y.; Sun, H.; Wu, L. Selenium inhibits ferroptosis and ameliorates autistic-like behaviors of BTBR mice by regulating the Nrf2/GPx4 pathway. Brain Res. Bull. 2022, 183, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Nordman, T.; Olsson, J.M.; Damdimopoulos, A.; Björkhem-Bergman, L.; Nalvarte, I.; Eriksson, L.C.; Arnér, E.S.; Spyrou, G.; Björnstedt, M. The mammalian cytosolic selenoenzyme thioredoxin reductase reduces ubiquinone. A novel mechanism for defense against oxidative stress. J. Biol. Chem. 2003, 278, 2141–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecorelli, A.; Ferrara, F.; Messano, N.; Cordone, V.; Schiavone, M.L.; Cervellati, F.; Woodby, B.; Cervellati, C.; Hayek, J.; Valacchi, G. Alterations of mitochondrial bioenergetics, dynamics, and morphology support the theory of oxidative damage involvement in autism spectrum disorder. Faseb J. 2020, 34, 6521–6538. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, S.; Singh, I.; Leung, W.; Stockler, S.; Ipsiroglu, O.S. Iron deficiency and common neurodevelopmental disorders—A scoping review. PLoS ONE 2022, 17, e0273819. [Google Scholar] [CrossRef]
- Gunes, S.; Ekinci, O.; Celik, T. Iron deficiency parameters in autism spectrum disorder: Clinical correlates and associated factors. Ital. J. Pediatr. 2017, 43, 86. [Google Scholar] [CrossRef]
- Glancy, B.; Kane, D.A.; Kavazis, A.N.; Goodwin, M.L.; Willis, W.T.; Gladden, L.B. Mitochondrial lactate metabolism: History and implications for exercise and disease. J. Physiol. 2021, 599, 863–888. [Google Scholar] [CrossRef]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [Green Version]
- Crane, F.L. Biochemical functions of coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef]
- Barcelos, I.P.; Haas, R.H. CoQ10 and Aging. Biology 2019, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Potgieter, M.; Pretorius, E.; Pepper, M.S. Primary and secondary coenzyme Q10 deficiency: The role of therapeutic supplementation. Nutr. Rev. 2013, 71, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorder: Unique Abnormalities and Targeted Treatments. Semin. Pediatr. Neurol. 2020, 35, 100829. [Google Scholar] [CrossRef] [PubMed]
- Mousavinejad, E.; Ghaffari, M.A.; Riahi, F.; Hajmohammadi, M.; Tiznobeyk, Z.; Mousavinejad, M. Coenzyme Q10 supplementation reduces oxidative stress and decreases antioxidant enzyme activity in children with autism spectrum disorders. Psychiatry Res. 2018, 265, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Gvozdjáková, A.; Kucharská, J.; Ostatníková, D.; Babinská, K.; Nakládal, D.; Crane, F.L. Ubiquinol improves symptoms in children with autism. Oxid. Med. Cell Longev. 2014, 2014, 798957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cucinotta, F.; Ricciardello, A.; Turriziani, L.; Mancini, A.; Keller, R.; Sacco, R.; Persico, A.M. Efficacy and Safety of Q10 Ubiquinol with Vitamins B and E in Neurodevelopmental Disorders: A Retrospective Chart Review. Front. Psychiatry 2022, 13, 829516. [Google Scholar] [CrossRef]
- El-Ansary, A.; Al-Ghamdi, M.; Bhat, R.S.; Al-Daihan, S.; Al-Ayadhi, L. Potency of pre-post treatment of coenzyme Q10 and melatonin supplement in ameliorating the impaired fatty acid profile in rodent model of autism. Food Nutr. Res. 2016, 60, 28127. [Google Scholar] [CrossRef] [Green Version]
- Rossignol, D.A.; Frye, R.E. Psychotropic Medications for Sleep Disorders in Autism Spectrum Disorder. In Handbook of Autism and Pervasive Developmental Disorders—Assessment, Diagnosis and Treatment; Malson, J.L., Sturmey, P., Eds.; Springer Nature: Cham, Switzerland, 2022. [Google Scholar]
- Failla, M.L.; Chitchumroonchokchai, C.; Aoki, F. Increased bioavailability of ubiquinol compared to that of ubiquinone is due to more efficient micellarization during digestion and greater GSH-dependent uptake and basolateral secretion by Caco-2 cells. J. Agric. Food Chem. 2014, 62, 7174–7182. [Google Scholar] [CrossRef]
- Langsjoen, P.H.; Langsjoen, A.M. Comparison study of plasma coenzyme Q10 levels in healthy subjects supplemented with ubiquinol versus ubiquinone. Clin. Pharmacol. Drug Dev. 2014, 3, 13–17. [Google Scholar] [CrossRef]
- Solmonson, A.; DeBerardinis, R.J. Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 2018, 293, 7522–7530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, S.M.; Romeiro, C.F.R.; Rodrigues, C.A.; Cerqueira, A.R.L.; Monteiro, M.C. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer’s Disease? Oxid. Med. Cell Longev. 2019, 2019, 8409329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, L.; Kraemer, K.; Rimbach, G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition 2001, 17, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Yang, X.; Cao, Y.; Long, X.; Shang, H.; Jia, Z. Role of lipoic acid in multiple sclerosis. CNS Neurosci. Ther. 2022, 28, 319–331. [Google Scholar] [CrossRef]
- Dragomanova, S.; Miteva, S.; Nicoletti, F.; Mangano, K.; Fagone, P.; Pricoco, S.; Staykov, H.; Tancheva, L. Therapeutic Potential of Alpha-Lipoic Acid in Viral Infections, including COVID-19. Antioxidants 2021, 10, 1294. [Google Scholar] [CrossRef]
- Gorąca, A.; Huk-Kolega, H.; Piechota, A.; Kleniewska, P.; Ciejka, E.; Skibska, B. Lipoic acid—Biological activity and therapeutic potential. Pharmacol. Rep. 2011, 63, 849–858. [Google Scholar] [CrossRef]
- Frye, R.E.; Huffman, L.C.; Elliott, G.R. Tetrahydrobiopterin as a novel therapeutic intervention for autism. Neurotherapeutics 2010, 7, 241–249. [Google Scholar] [CrossRef]
- Thöny, B.; Auerbach, G.; Blau, N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 2000, 347, 1–16. [Google Scholar] [CrossRef]
- Klaiman, C.; Huffman, L.; Masaki, L.; Elliott, G.R. Tetrahydrobiopterin as a treatment for autism spectrum disorders: A double-blind, placebo-controlled trial. J. Child Adolesc. Psychopharmacol. 2013, 23, 320–328. [Google Scholar] [CrossRef]
- Frye, R.E.; DeLatorre, R.; Taylor, H.B.; Slattery, J.; Melnyk, S.; Chowdhury, N.; James, S.J. Metabolic effects of sapropterin treatment in autism spectrum disorder: A preliminary study. Transl. Psychiatry 2013, 3, e237. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Indika, N.-L.R.; Frye, R.E.; Rossignol, D.A.; Owens, S.C.; Senarathne, U.D.; Grabrucker, A.M.; Perera, R.; Engelen, M.P.K.J.; Deutz, N.E.P. The Rationale for Vitamin, Mineral, and Cofactor Treatment in the Precision Medical Care of Autism Spectrum Disorder. J. Pers. Med. 2023, 13, 252. https://doi.org/10.3390/jpm13020252
Indika N-LR, Frye RE, Rossignol DA, Owens SC, Senarathne UD, Grabrucker AM, Perera R, Engelen MPKJ, Deutz NEP. The Rationale for Vitamin, Mineral, and Cofactor Treatment in the Precision Medical Care of Autism Spectrum Disorder. Journal of Personalized Medicine. 2023; 13(2):252. https://doi.org/10.3390/jpm13020252
Chicago/Turabian StyleIndika, Neluwa-Liyanage R., Richard E. Frye, Daniel A. Rossignol, Susan C. Owens, Udara D. Senarathne, Andreas M. Grabrucker, Rasika Perera, Marielle P. K. J. Engelen, and Nicolaas E. P. Deutz. 2023. "The Rationale for Vitamin, Mineral, and Cofactor Treatment in the Precision Medical Care of Autism Spectrum Disorder" Journal of Personalized Medicine 13, no. 2: 252. https://doi.org/10.3390/jpm13020252