
Management of Hyponatremia in Heart Failure: Practical Considerations
 ,
,  , , , , , and
, , , , , and
Abstract
:1. Introduction
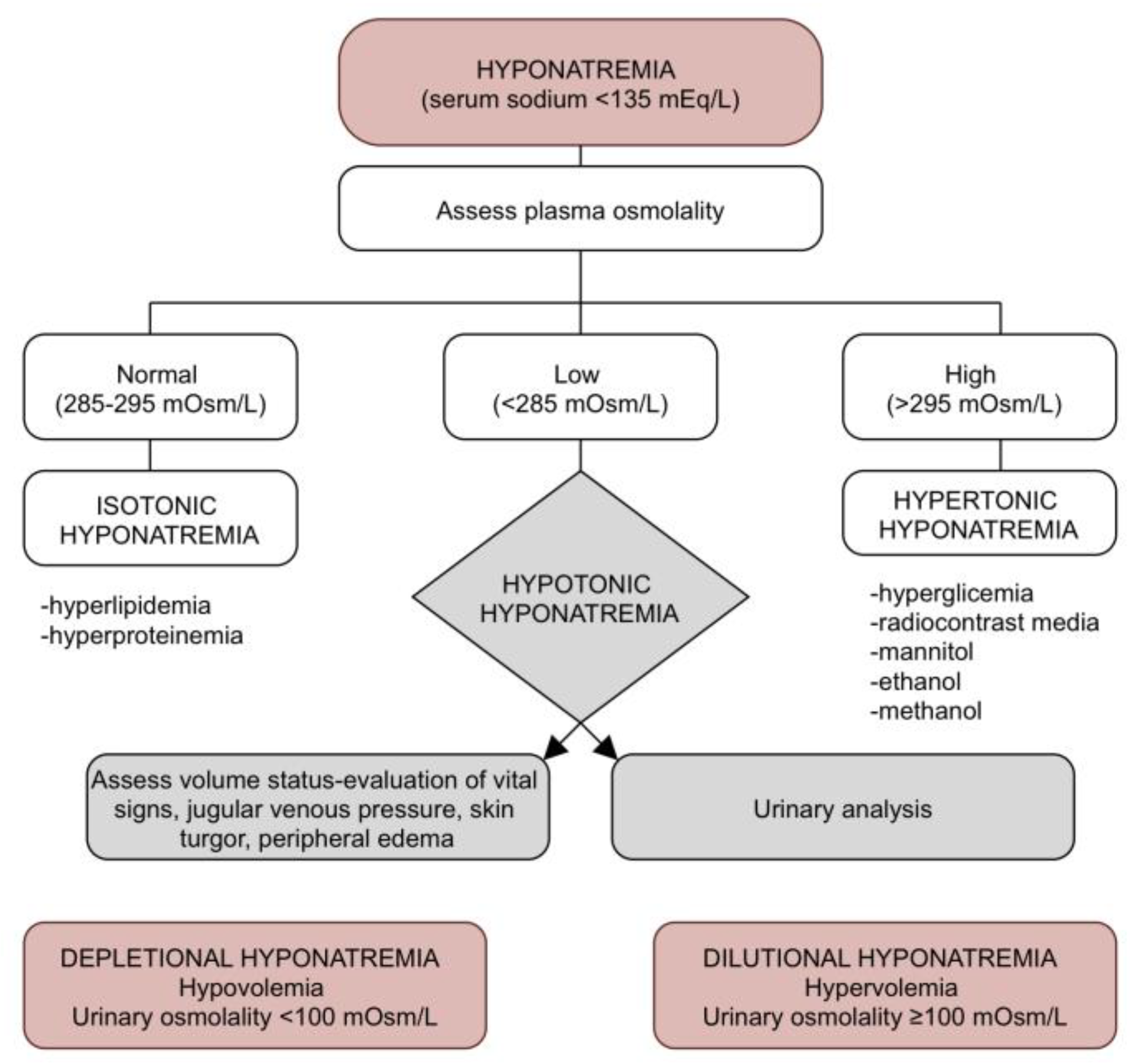
2. Dilutional Hyponatremia
3. Depletional Hyponatremia
4. Clinical Aspects in Hyponatremia
5. Diagnosis of Heart Failure Associated Hyponatremia
6. Treatment of Acute and Severe Hyponatremia
7. Treatment of Dilutional Hyponatremia
7.1. Loop Diuretics
7.2. Hypertonic Saline Solution
7.3. Fluid Restriction
7.4. Vaptans
7.5. Other Therapy Options
7.6. Ultrafiltration
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adrogué, H.J. Hyponatremia in Heart Failure. Methodist DeBakey Cardiovasc. J. 2017, 13, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheorghiade, M.; Abraham, W.T.; Albert, N.M.; Gattis Stough, W.; Greenberg, B.H.; O’Connor, C.M.; She, L.; Yancy, C.W.; Young, J.; Fonarow, G.C. Relationship between Admission Serum Sodium Concentration and Clinical Outcomes in Patients Hospitalized for Heart Failure: An Analysis from the OPTIMIZE-HF Registry. Eur. Heart J. 2007, 28, 980–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, M.; Haraguchi, G.; Ohigashi, H.; Sasaoka, T.; Yoshikawa, S.; Inagaki, H.; Ashikaga, T.; Isobe, M. Progression of Hyponatremia Is Associated with Increased Cardiac Mortality in Patients Hospitalized for Acute Decompensated Heart Failure. J. Card. Fail. 2012, 18, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Shchekochikhin, D.Y.; Schrier, R.W.; Lindenfeld, J.; Price, L.L.; Jaber, B.L.; Madias, N.E. Outcome Differences in Community- versus Hospital-Acquired Hyponatremia in Patients with a Diagnosis of Heart Failure. Circ. Heart Fail. 2013, 6, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheorghiade, M.; Rossi, J.S.; Cotts, W.; Shin, D.D.; Hellkamp, A.S.; Piña, I.L.; Fonarow, G.C.; DeMarco, T.; Pauly, D.F.; Rogers, J.; et al. Characterization and Prognostic Value of Persistent Hyponatremia in Patients with Severe Heart Failure in the ESCAPE Trial. Arch. Intern. Med. 2007, 167, 1998–2005. [Google Scholar] [CrossRef]
- Omar, H.R.; Charnigo, R.; Guglin, M. Prognostic Significance of Discharge Hyponatremia in Heart Failure Patients with Normal Admission Sodium (from the ESCAPE Trial). Am. J. Cardiol. 2017, 120, 607–615. [Google Scholar] [CrossRef]
- Rodriguez, M.; Hernandez, M.; Cheungpasitporn, W.; Kashani, K.B.; Riaz, I.; Rangaswami, J.; Herzog, E.; Guglin, M.; Krittanawong, C. Hyponatremia in Heart Failure: Pathogenesis and Management. Curr. Cardiol. Rev. 2019, 15, 252–261. [Google Scholar] [CrossRef]
- Sica, D.A. Sodium and Water Retention in Heart Failure and Diuretic Therapy: Basic Mechanisms. Cleve. Clin. J. Med. 2006, 73, S2. [Google Scholar] [CrossRef]
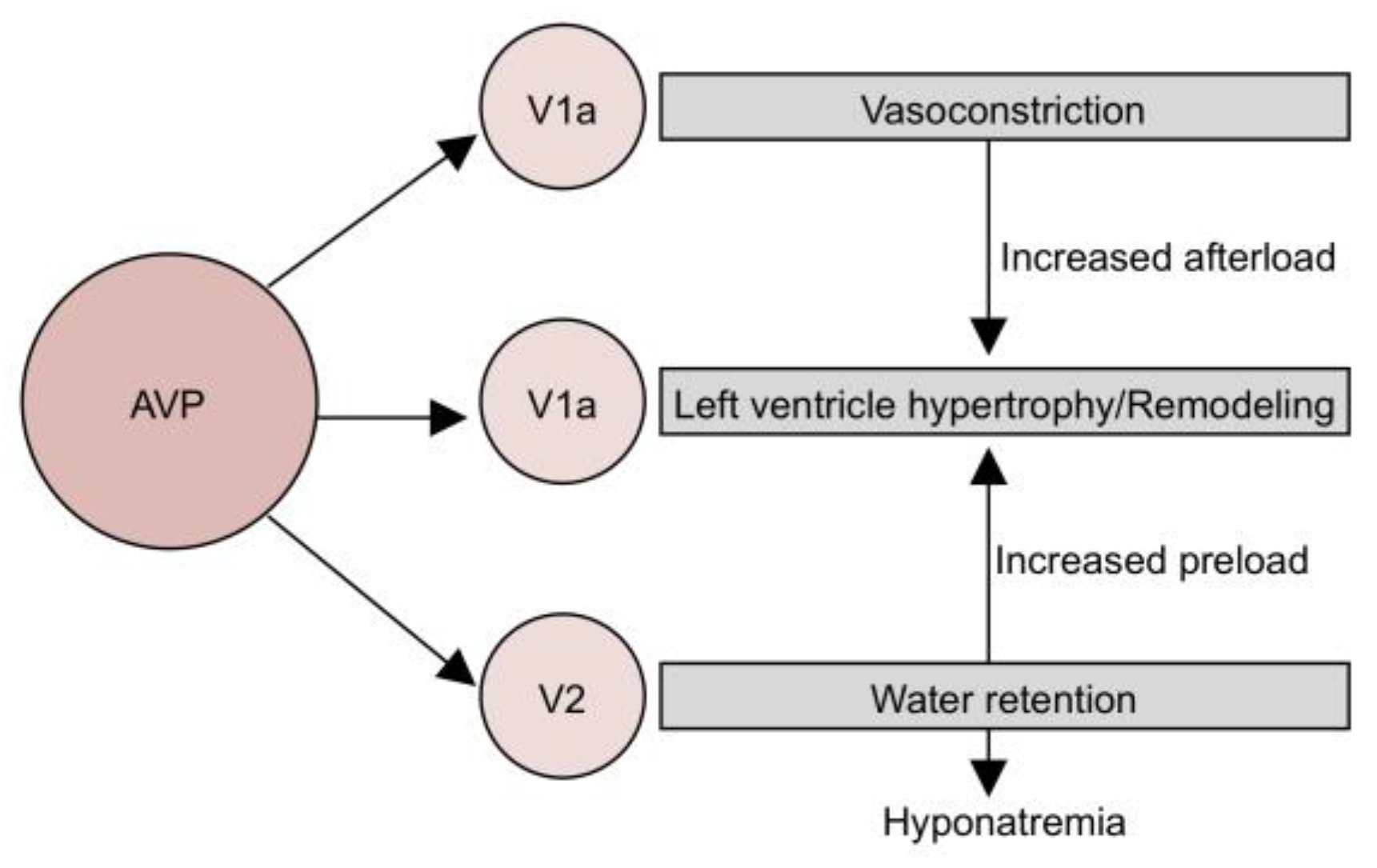
- Bankir, L.; Bichet, D.G.; Morgenthaler, N.G. Vasopressin: Physiology, Assessment and Osmosensation. J. Intern. Med. 2017, 282, 284–297. [Google Scholar] [CrossRef] [Green Version]
- LeJemtel, T.H.; Serrano, C. Vasopressin Dysregulation: Hyponatremia, Fluid Retention and Congestive Heart Failure. Int. J. Cardiol. 2007, 120, 1–9. [Google Scholar] [CrossRef]
- Benedict, C.R.; Weiner, D.H.; Johnstone, D.E.; Bourassa, M.G.; Ghali, J.K.; Nicklas, J.; Kirlin, P.; Greenberg, B.; Quinones, M.A.; Yusuf, S. Comparative Neurohormonal Responses in Patients with Preserved and Impaired Left Ventricular Ejection Fraction: Results of the Studies of Left Ventricular Dysfunction (SOLVD) Registry. The SOLVD Investigators. J. Am. Coll. Cardiol. 1993, 22, 146A–153A. [Google Scholar] [CrossRef] [Green Version]
- Ranieri, M.; Di Mise, A.; Tamma, G.; Valenti, G. Vasopressin—Aquaporin-2 Pathway: Recent Advances in Understanding Water Balance Disorders. F1000Research 2019, 8, 149. [Google Scholar] [CrossRef] [Green Version]
- Aoyagi, T.; Koshimizu, T.; Tanoue, A. Vasopressin Regulation of Blood Pressure and Volume: Findings from V1a Receptor—Deficient Mice. Kidney Int. 2009, 76, 1035–1039. [Google Scholar] [CrossRef] [Green Version]
- Hiroyama, M.; Wang, S.; Aoyagi, T.; Oikawa, R.; Sanbe, A.; Takeo, S.; Tanoue, A. Vasopressin Promotes Cardiomyocyte Hypertrophy via the Vasopressin V1A Receptor in Neonatal Mice. Eur. J. Pharmacol. 2007, 559, 89–97. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 79, 1757–1780. [Google Scholar] [CrossRef]
- Kennelly, P.; Sapkota, R.; Azhar, M.; Cheema, F.H.; Conway, C.; Hameed, A. Diuretic Therapy in Congestive Heart Failure. Acta Cardiol. 2021, 77, 97–104. [Google Scholar] [CrossRef]
- Ellison, D.H. Clinical Pharmacology in Diuretic Use. Clin. J. Am. Soc. Nephrol. 2019, 14, 1248–1257. [Google Scholar] [CrossRef] [Green Version]
- Shams, E.; Bonnice, S.; Mayrovitz, H.N. Diuretic Resistance Associated with Heart Failure. Cureus 2022, 14, e21369. [Google Scholar] [CrossRef]
- Kiernan, M.S.; Stevens, S.R.; Tang, W.H.W.; Butler, J.; Anstrom, K.J.; Birati, E.Y.; Grodin, J.L.; Gupta, D.; Margulies, K.B.; LaRue, S.; et al. Determinants of Diuretic Responsiveness and Associated Outcomes during Acute Heart Failure Hospitalization: An Analysis from the NHLBI Heart Failure Network Clinical Trials. J. Card. Fail. 2018, 24, 428–438. [Google Scholar] [CrossRef]
- Kaissling, B.; Stanton, B.A. Adaptation of Distal Tubule and Collecting Duct to Increased Sodium Delivery. I. Ultrastructure. Am. J. Physiol. 1988, 255, F1256–F1268. [Google Scholar] [CrossRef]
- Jentzer, J.C.; DeWald, T.A.; Hernandez, A.F. Combination of Loop Diuretics with Thiazide-Type Diuretics in Heart Failure. J. Am. Coll. Cardiol. 2010, 56, 1527–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, K.S.; Kim, G.-H. Thiazide-Induced Hyponatremia. Electrolyte Blood Press. 2010, 8, 51. [Google Scholar] [CrossRef] [Green Version]
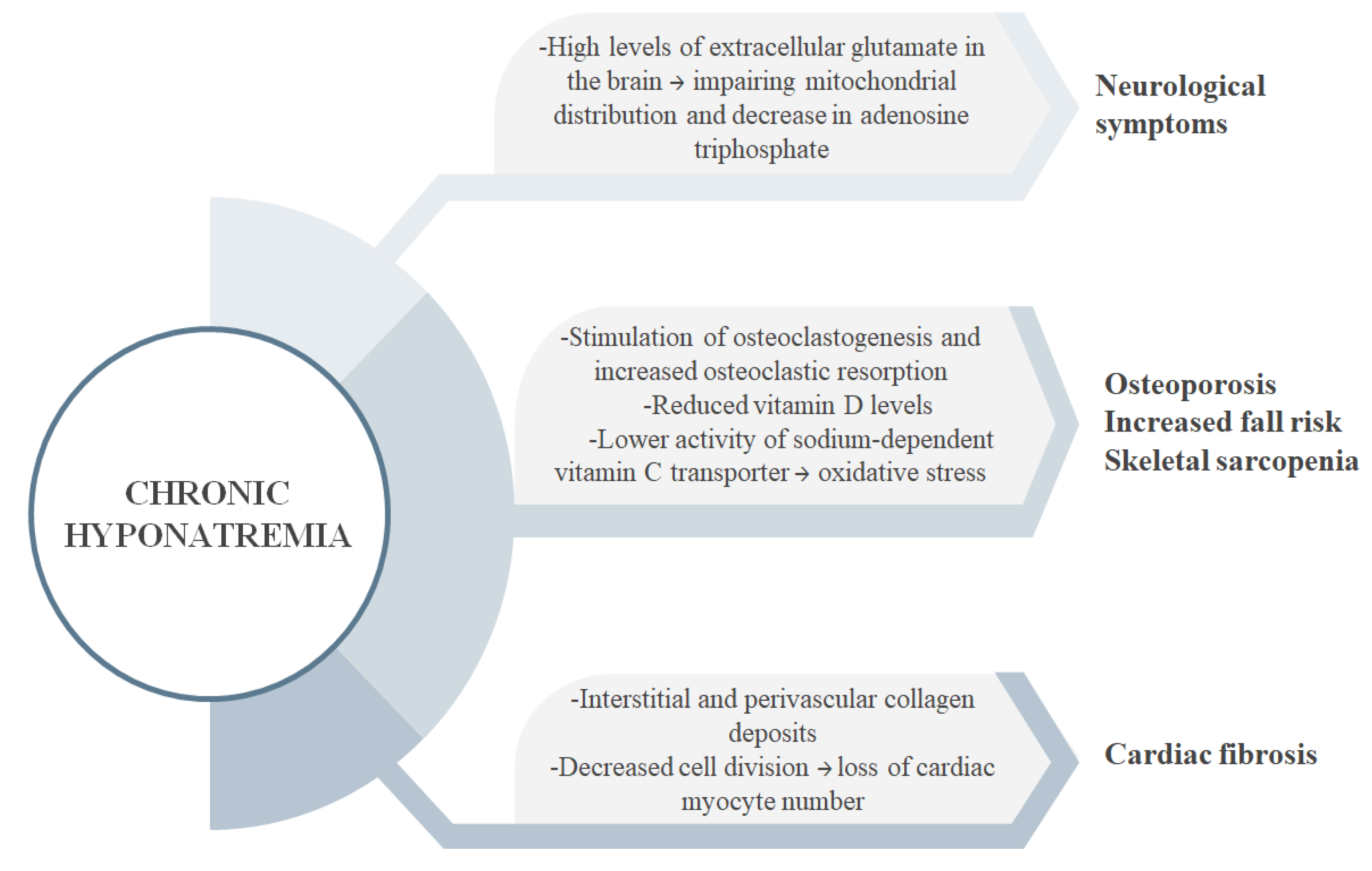
- Giuliani, C.; Peri, A. Effects of Hyponatremia on the Brain. J. Clin. Med. 2014, 3, 1163–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbalis, J.G.; Goldsmith, S.R.; Greenberg, A.; Korzelius, C.; Schrier, R.W.; Sterns, R.H.; Thompson, C.J. Diagnosis, Evaluation, and Treatment of Hyponatremia: Expert Panel Recommendations. Am. J. Med. 2013, 126, S1–S42. [Google Scholar] [CrossRef] [PubMed]
- Ayus, J.C.; Achinger, S.G.; Arieff, A. Brain Cell Volume Regulation in Hyponatremia: Role of Sex, Age, Vasopressin, and Hypoxia. Am. J. Physiol. Renal. Physiol. 2008, 295, F619–F624. [Google Scholar] [CrossRef]
- Verbalis, J.G. Brain Volume Regulation in Response to Changes in Osmolality. Neuroscience 2010, 168, 862–870. [Google Scholar] [CrossRef]
- Pasantes-Morales, H.; Franco, R.; Ordaz, B.; Ochoa, L.D. Mechanisms Counteracting Swelling in Brain Cells during Hyponatremia. Arch. Med. Res. 2002, 33, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, H.; Sugimura, Y.; Takagi, H.; Mizoguchi, H.; Takeuchi, H.; Izumida, H.; Nakashima, K.; Ochiai, H.; Takeuchi, S.; Kiyota, A.; et al. Chronic Hyponatremia Causes Neurologic and Psychologic Impairments. J. Am. Soc. Nephrol. 2016, 27, 766–780. [Google Scholar] [CrossRef] [Green Version]
- Renneboog, B.; Musch, W.; Vandemergel, X.; Manto, M.U.; Decaux, G. Mild Chronic Hyponatremia Is Associated with Falls, Unsteadiness, and Attention Deficits. Am. J. Med. 2006, 119, 71.e1–71.e8. [Google Scholar] [CrossRef]
- Jäckle, K.; Klockner, F.; Hoffmann, D.B.; Roch, P.J.; Reinhold, M.; Lehmann, W.; Weiser, L. Influence of Hyponatremia on Spinal Bone Quality and Fractures due to Low-Energy Trauma. Medicina 2021, 57, 1224. [Google Scholar] [CrossRef]
- Barsony, J.; Sugimura, Y.; Verbalis, J.G. Osteoclast Response to Low Extracellular Sodium and the Mechanism of Hyponatremia-Induced Bone Loss. J. Biol. Chem. 2010, 286, 10864–10875. [Google Scholar] [CrossRef] [Green Version]
- Barsony, J.; Manigrasso, M.B.; Xu, Q.; Tam, H.; Verbalis, J.G. Chronic Hyponatremia Exacerbates Multiple Manifestations of Senescence in Male Rats. Age 2012, 35, 271–288. [Google Scholar] [CrossRef] [Green Version]
- Danbolt, N.C. Glutamate Uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Verbalis, J.G.; Gullans, S.R. Hyponatremia Causes Large Sustained Reductions in Brain Content of Multiple Organic Osmolytes in Rats. Brain Res. 1991, 567, 274–282. [Google Scholar] [CrossRef]
- Takeuchi, H.; Mizuno, T.; Zhang, G.; Wang, J.; Kawanokuchi, J.; Kuno, R.; Suzumura, A. Neuritic Beading Induced by Activated Microglia Is an Early Feature of Neuronal Dysfunction toward Neuronal Death by Inhibition of Mitochondrial Respiration and Axonal Transport. J. Biol. Chem. 2005, 280, 10444–10454. [Google Scholar] [CrossRef] [Green Version]
- Tandukar, S.; Sterns, R.H.; Rondon-Berrios, H. Osmotic Demyelination Syndrome Following Correction of Hyponatremia by ≤10 MEq/L per Day. Kidney360 2021, 2, 1415–1423. [Google Scholar] [CrossRef]
- Elshafei, M.; Danjuma, M.I.; Tahir, R.E. Therapeutic Challenges in the Management of Osmotic Demyelination Syndrome: A Case Report of a Favorable Outcome from a Tertiary Center. Medicine 2020, 99, e20283. [Google Scholar] [CrossRef]
- Garg, P.; Aggarwal, A.; Malhotra, R.; Dhall, S. Osmotic Demyelination Syndrome-Evolution of Extrapontine before Pontine Myelinolysis on Magnetic Resonance Imaging. J. Neurosci. Rural Pract. 2019, 10, 126–135. [Google Scholar] [CrossRef] [Green Version]
- King, J.D.; Rosner, M.H. Osmotic Demyelination Syndrome. Am. J. Med. Sci. 2010, 339, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Spasovski, G.; Vanholder, R.; Allolio, B.; Annane, D.; Ball, S.; Bichet, D.; Decaux, G.; Fenske, W.; Hoorn, E.J.; Ichai, C.; et al. Clinical Practice Guideline on Diagnosis and Treatment of Hyponatraemia. Eur. J. Endocrinol. 2014, 170, G1–G47. [Google Scholar] [CrossRef]
- Lippi, G.; Aloe, R. Hyponatremia and Pseudohyponatremia: First, Do No Harm. Am. J. Med. 2010, 123, e17. [Google Scholar] [CrossRef] [PubMed]
- Tzamaloukas, A.H.; Khitan, Z.J.; Glew, R.H.; Roumelioti, M.; Rondon-Berrios, H.; Elisaf, M.S.; Raj, D.S.; Owen, J.; Sun, Y.; Siamopoulos, K.C.; et al. Serum Sodium Concentration and Tonicity in Hyperglycemic Crises: Major Influences and Treatment Implications. J. Am. Heart. Assoc. 2019, 8, e011786. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, F.H.; Steels, P.; Grieten, L.; Nijst, P.; Tang, W.H.W.; Mullens, W. Hyponatremia in Acute Decompensated Heart Failure: Depletion versus Dilution. J. Am. Coll. Cardiol. 2015, 65, 480–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterns, R.H. Treatment of Severe Hyponatremia. Clin. J. Am. Soc. Nephrol. 2018, 13, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoorn, E.J.; Zietse, R. Diagnosis and Treatment of Hyponatremia: Compilation of the Guidelines. J. Am. Soc. Nephrol. 2017, 28, 1340–1349. [Google Scholar] [CrossRef] [Green Version]
- Ijaiya, T.; Manohar, S.; Lakshmi, K. Therapeutic Approach to the Management of Severe Asymptomatic Hyponatremia. Case Rep. Nephrol. 2017, 2017, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Sirbu, O.; Sorodoc, V.; Jaba, I.M.; Floria, M.; Stoica, A.; Profire, L.; Tuchilus, C.; Rusu, G.; Sorodoc, L. The Influence of Cardiovascular Medications on Iron Metabolism in Patients with Heart Failure. Medicina 2019, 55, 329. [Google Scholar] [CrossRef] [Green Version]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Stoica, A.; Şorodoc, V.; Lionte, C.; Jaba, I.M.; Costache, I.; Anisie, E.; Tuchiluș, C.; Rusalim Petriș, O.; Sîrbu, O.; Jaba, E.; et al. Acute Cardiac Dyspnea in the Emergency Department: Diagnostic Value of N-Terminal Prohormone of Brain Natriuretic Peptide and Galectin-3. J. Int. Med. Res. 2019, 47, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Grewal, G.S.; Kozik, T.M.; Hickman, M.C.; Connolly, T.F.; Bajwa, R.S.; Bhattacharyya, M. Feasibility Study to Determine If Administration of Carbonic Anhydrase Inhibitor Acetazolamide (Diamox) Will Cause Aquaresis but Prevent Hyponatriemia in a Healthy Population: SALT Study. J. Integr. Cardiol. 2018, 4, 1–3. [Google Scholar] [CrossRef]
- Damman, K.; Testani, J.M. The Kidney in Heart Failure: An Update. Eur. Heart. J. 2015, 36, 1437–1444. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.L.; Borgeson, D.D.; Grantham, J.A.; Luchner, A.; Redfield, M.M.; Burnett, J.C. Dietary Sodium Modulation of Aldosterone Activation and Renal Function during the Progression of Experimental Heart Failure. Eur. J. Heart. Fail. 2014, 17, 144–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttolomondo, A.; Maida, C.; Casuccio, A.; Di Raimondo, D.; Fonte, R.; Vassallo, V.; Puleo, M.G.; Di Chiara, T.; Mogavero, A.; Del Cuore, A.; et al. Effects of Intravenous Furosemide plus Small-Volume Hypertonic Saline Solutions on Markers of Heart Failure. ESC Heart Fail. 2021, 8, 4174–4186. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.; Soufer, A.; Goljo, E.; Colna, M.; Rao, V.S.; Jeon, S.; Raghavendra, P.; D’Ambrosi, J.; Riello, R.; Coca, S.G.; et al. Real World Use of Hypertonic Saline in Refractory Acute Decompensated Heart Failure. JACC Heart Fail. 2020, 8, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Li, L.; Niu, H.; Ma, X.; Yang, J.; Yuan, C.; Mu, G.; Zhang, J. Impact of Compound Hypertonic Saline Solution on Decompensated Heart Failure. Int. Heart. J. 2017, 58, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Lafrenière, G.; Béliveau, P.; Bégin, J.-Y.; Simonyan, D.; Côté, S.; Gaudreault, V.; Israeli, Z.; Lavi, S.; Bagur, R. Effects of Hypertonic Saline Solution on Body Weight and Serum Creatinine in Patients with Acute Decompensated Heart Failure. World. J. Cardiol. 2017, 9, 685. [Google Scholar] [CrossRef]
- Yayla, Ç.; Akyel, A.; Canpolat, U.; Gayretli Yayla, K.; Eyiol, A.; Akboğa, M.K.; Türkoğlu, S.; Tavil, Y.; Boyacı, B.; Çengel, A. Comparison of Three Diuretic Treatment Strategies for Patients with Acute Decompensated Heart Failure. Herz 2015, 40, 1115–1120. [Google Scholar] [CrossRef]
- Paterna, S.; Di Gaudio, F.; La Rocca, V.; Balistreri, F.; Greco, M.; Torres, D.; Lupo, U.; Rizzo, G.; di Pasquale, P.; Indelicato, S.; et al. Hypertonic Saline in Conjunction with High-Dose Furosemide Improves Dose–Response Curves in Worsening Refractory Congestive Heart Failure. Adv. Ther. 2015, 32, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Okuhara, Y.; Hirotani, S.; Naito, Y.; Nakabo, A.; Iwasaku, T.; Eguchi, A.; Morisawa, D.; Ando, T.; Sawada, H.; Manabe, E.; et al. Intravenous Salt Supplementation with Low-Dose Furosemide for Treatment of Acute Decompensated Heart Failure. J. Card. Fail. 2014, 20, 295–301. [Google Scholar] [CrossRef]
- Issa, V.S.; Andrade, L.; Ayub-Ferreira, S.M.; Bacal, F.; de Bragança, A.C.; Guimarães, G.V.; Marcondes-Braga, F.G.; Cruz, F.D.; Chizzola, P.R.; Conceição-Souza, G.E.; et al. Hypertonic Saline Solution for Prevention of Renal Dysfunction in Patients with Decompensated Heart Failure. Int. J. Cardiol. 2013, 167, 34–40. [Google Scholar] [CrossRef]
- Parrinello, G.; Di Pasquale, P.; Torres, D.; Cardillo, M.; Schimmenti, C.; Lupo, U.; Iatrino, R.; Petrantoni, R.; Montaina, C.; Giambanco, S.; et al. Troponin I Release after Intravenous Treatment with High Furosemide Doses plus Hypertonic Saline Solution in Decompensated Heart Failure Trial (Tra-HSS-Fur). Am. Heart. J. 2012, 164, 351–357. [Google Scholar] [CrossRef]
- Parrinello, G.; Paterna, S.; Di Pasquale, P.; Torres, D.; Mezzero, M.; Cardillo, M.; Fasullo, S.; La Rocca, G.; Licata, G. Changes in Estimating Echocardiography Pulmonary Capillary Wedge Pressure after Hypersaline plus Furosemide versus Furosemide Alone in Decompensated Heart Failure. J. Card. Fail. 2011, 17, 331–339. [Google Scholar] [CrossRef]
- Paterna, S.; Fasullo, S.; Cannizzaro, S.; Vitrano, G.; Terrazzino, G.; Maringhini, G.; Ganci, F.; Scalzo, S.; Di Pasquale, P.; Parrinello, G.; et al. Short-Term Effects of Hypertonic Saline Solution in Acute Heart Failure and Long-Term Effects of a Moderate Sodium Restriction in Patients with Compensated Heart Failure with New York Heart Association Class III (Class C) (SMAC-HF Study). Am. J. Med. Sci. 2011, 342, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Paterna, S.; Di Pasquale, P.; Parrinello, G.; Fornaciari, E.; Di Gaudio, F.; Fasullo, S.; Giammanco, M.; Sarullo, F.M.; Licata, G. Changes in Brain Natriuretic Peptide Levels and Bioelectrical Impedance Measurements after Treatment with High-Dose Furosemide and Hypertonic Saline Solution versus High-Dose Furosemide Alone in Refractory Congestive Heart Failure. J. Am. Coll. Cardiol. 2005, 45, 1997–2003. [Google Scholar] [CrossRef] [Green Version]
- Licata, G.; Pasquale, P.D.; Parrinello, G.; Cardinale, A.; Scandurra, A.; Follone, G.; Argano, C.; Tuttolomondo, A.; Paterna, S. Effects of High-Dose Furosemide and Small-Volume Hypertonic Saline Solution Infusion in Comparison with a High Dose of Furosemide as Bolus in Refractory Congestive Heart Failure: Long-Term Effects. Am. Heart. J. 2003, 145, 459–466. [Google Scholar] [CrossRef]
- Paterna, S.; Di Pasquale, P.; Parrinello, G.; Amato, P.; Cardinale, A.; Follone, G.; Giubilato, A.; Licata, G. Effects of High-Dose Furosemide and Small-Volume Hypertonic Saline Solution Infusion in Comparison with a High Dose of Furosemide as a Bolus, in Refractory Congestive Heart Failure. Eur. J. Heart Fail. 2000, 2, 305–313. [Google Scholar] [CrossRef]
- Dunlap, M.E.; Hauptman, P.J.; Amin, A.N.; Chase, S.L.; Chiodo, J.A.; Chiong, J.R.; Dasta, J.F. Current Management of Hyponatremia in Acute Heart Failure: A Report from the Hyponatremia Registry for Patients with Euvolemic and Hypervolemic Hyponatremia (HN Registry). J. Am. Heart Assoc. 2017, 6, e005261. [Google Scholar] [CrossRef]
- Eng, S.H.; Jaarsma, T.; Lupón, J.; González, B.; Ehrlin, J.; Díaz, V.; Bayes-Genis, A.; Waldréus, N. Thirst and Factors Associated with Frequent Thirst in Patients with Heart Failure in Spain. Heart Lung 2021, 50, 86–91. [Google Scholar] [CrossRef]
- Herrmann, J.J.; Beckers-Wesche, F.; Baltussen, L.E.H.J.M.; Verdijk, M.H.I.; Bellersen, L.; Brunner-la Rocca, H.-P.; Jaarsma, T.; Pisters, R.; Sanders-van Wijk, S.; Rodwell, L.; et al. Fluid REStriction in Heart Failure vs Liberal Fluid UPtake: Rationale and Design of the Randomized FRESH-up Study. J. Card. Fail. 2022, 28, 1522–1530. [Google Scholar] [CrossRef]
- Finley, J.J.; Konstam, M.A.; Udelson, J.E. Arginine Vasopressin Antagonists for the Treatment of Heart Failure and Hyponatremia. Circulation 2008, 118, 410–421. [Google Scholar] [CrossRef]
- Narayen, G.; Mandal, S. Vasopressin Receptor Antagonists and Their Role in Clinical Medicine. J. Endocrinol. Metab. 2012, 16, 183. [Google Scholar] [CrossRef] [PubMed]
- Smetana, K.S.; Wiss, A.L.; May, C.C. Efficacy and Safety of Conivaptan versus Tolvaptan in Neurocritically Ill Patients. J. Transl. Crit. Care Med. 2022, 4, 7. [Google Scholar] [CrossRef]
- Decaux, G.; Soupart, A.; Vassart, G. Non-Peptide Arginine-Vasopressin Antagonists: The Vaptans. Lancet 2008, 371, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Udelson, J.E.; Bilsker, M.; Hauptman, P.J.; Sequeira, R.; Thomas, I.; O’Brien, T.; Zimmer, C.; Orlandi, C.; Konstam, M.A. A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study of Tolvaptan Monotherapy Compared to Furosemide and the Combination of Tolvaptan and Furosemide in Patients with Heart Failure and Systolic Dysfunction. J. Cardiol. Fail. 2011, 17, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.M.H.; Grazette, L.P.; Fong, M.W.; Yoon, A.J.; Lou, M.; Kuo, A.; Upadhyay, R.Y.; Han, E.E.; Mehra, A.; Elkayam, U. Tolvaptan vs. Furosemide-Based Diuretic Regimens in Patients Hospitalized for Heart Failure with Hyponatremia (AQUA-AHF). ESC Heart Fail. 2020, 7, 1927–1934. [Google Scholar] [CrossRef]
- Schrier, R.W.; Gross, P.; Gheorghiade, M.; Berl, T.; Verbalis, J.G.; Czerwiec, F.S.; Orlandi, C. Tolvaptan, a Selective Oral Vasopressin V2-Receptor Antagonist, for Hyponatremia. N. Engl. J. Med. 2006, 355, 2099–2112. [Google Scholar] [CrossRef] [Green Version]
- Konstam, M.A. Effects of Oral Tolvaptan in Patients Hospitalized for Worsening Heart Failurethe EVEREST Outcome Trial. JAMA 2007, 297, 1319. [Google Scholar] [CrossRef]
- Felker, G.M.; Mentz, R.J.; Cole, R.T.; Adams, K.F.; Egnaczyk, G.F.; Fiuzat, M.; Patel, C.B.; Echols, M.; Khouri, M.G.; Tauras, J.M.; et al. Efficacy and Safety of Tolvaptan in Patients Hospitalized with Acute Heart Failure. J. Am. College Card. 2017, 69, 1399–1406. [Google Scholar] [CrossRef]
- Rossi, J.; Bayram, M.; Udelson, J.E.; Lloyd-Jones, D.; Adams, K.F.; Oconnor, C.M.; Stough, W.G.; Ouyang, J.; Shin, D.D.; Orlandi, C.; et al. Improvement in Hyponatremia during Hospitalization for Worsening Heart Failure Is Associated with Improved Outcomes: Insights from the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Chronic Heart Failure (ACTIV in CHF) Trial. Acute Card. Care 2007, 9, 82–86. [Google Scholar] [CrossRef]
- Konstam, M.A.; Kiernan, M.; Chandler, A.; Dhingra, R.; Mody, F.V.; Eisen, H.; Haught, W.H.; Wagoner, L.; Gupta, D.; Patten, R.; et al. Short-Term Effects of Tolvaptan in Patients with Acute Heart Failure and Volume Overload. J. Am. Coll. Cardiol. 2017, 69, 1409–1419. [Google Scholar] [CrossRef]
- Urbach, J.; Goldsmith, S.R. Vasopressin Antagonism in Heart Failure: A Review of the Hemodynamic Studies and Major Clinical Trials. Ther. Adv. Cardiovasc. Dis. 2021, 15, 175394472097774. [Google Scholar] [CrossRef]
- Schrier, R.W.; Sharma, S.; Shchekochikhin, D. Hyponatraemia: More than Just a Marker of Disease Severity? Nat. Rev. Nephrol. 2012, 9, 37–50. [Google Scholar] [CrossRef]
- Ghali, J.; Zmily, H.; Daifallah, S. Tolvaptan, Hyponatremia, and Heart Failure. Int. J. Nephrol. Renovasc. Dis. 2011, 4, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Aditya, S.; Rattan, A. Vaptans: A New Option in the Management of Hyponatremia. Int. J. Appl. Basic Med. Res. 2012, 2, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Schumann, J.; Henrich, E.C.; Strobl, H.; Prondzinsky, R.; Weiche, S.; Thiele, H.; Werdan, K.; Frantz, S.; Unverzagt, S. Inotropic Agents and Vasodilator Strategies for the Treatment of Cardiogenic Shock or Low Cardiac Output Syndrome. Cochrane Database Syst. Rev. 2018, 1, CD009669. [Google Scholar] [CrossRef]
- Mullens, W.; Abrahams, Z.; Francis, G.S.; Skouri, H.N.; Starling, R.C.; Young, J.B.; Taylor, D.O.; Tang, W.H.W. Sodium Nitroprusside for Advanced Low-Output Heart Failure. J. Am. Coll. Cardiol. 2008, 52, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Mullens, W.; Abrahams, Z.; Francis, G.S.; Sokos, G.; Starling, R.C.; Young, J.B.; Taylor, D.O.; Tang, W.H.W. Usefulness of Isosorbide Dinitrate and Hydralazine as Add-on Therapy in Patients Discharged for Advanced Decompensated Heart Failure. Am. J. Cardiol. 2009, 103, 1113–1119. [Google Scholar] [CrossRef] [Green Version]
- Balling, L.; Kober, L.; Schou, M.; Torp-Pedersen, C.; Gustafsson, F. Efficacy and Safety of Angiotensin-Converting Enzyme Inhibitors in Patients with Left Ventricular Systolic Dysfunction and Hyponatremia. J. Card. Fail. 2013, 19, 725–730. [Google Scholar] [CrossRef]
- Somaschini, A.; Casirati, A.; Cornara, S.; Demarchi, A.; Mandurino-Mirizzi, A.; Androulakis, E.; Lioudaki, E. Extracorporeal Veno-Venous Ultrafiltration in Patients with Acute Heart Failure. Rev. Cardiovasc. Med. 2021, 22, 1311. [Google Scholar] [CrossRef]
- Emani, S. Ultrafiltration for the Treatment of Acute Heart Failure. Heart Fail. Clin. 2018, 14, 517–524. [Google Scholar] [CrossRef]
- Kazory, A. Ultrafiltration Therapy for Heart Failure: Balancing Likely Benefits against Possible Risks. Clin. J. Am. Soc. Nephrol. 2016, 11, 1463–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costanzo, M.R.; Negoianu, D.; Jaski, B.E.; Bart, B.A.; Heywood, J.T.; Anand, I.S.; Smelser, J.M.; Kaneshige, A.M.; Chomsky, D.B.; Adler, E.D.; et al. Aquapheresis versus Intravenous Diuretics and Hospitalizations for Heart Failure. JACC. Heart Fail. 2016, 4, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Marenzi, G.; Muratori, M.; Cosentino, E.R.; Rinaldi, E.R.; Donghi, V.; Milazzo, V.; Ferramosca, E.; Borghi, C.; Santoro, A.; Agostoni, P. Continuous Ultrafiltration for Congestive Heart Failure: The CUORE Trial. J. Card. Fail. 2014, 20, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bart, B.A.; Goldsmith, S.R.; Lee, K.L.; Redfield, M.M.; Felker, G.M.; O’Connor, C.M.; Chen, H.H.; Rouleau, J.L.; Givertz, M.M.; Semigran, M.J.; et al. Cardiorenal Rescue Study in Acute Decompensated Heart Failure: Rationale and Design of CARRESS-HF, for the Heart Failure Clinical Research Network. J. Card. Fail. 2012, 18, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Giglioli, C.; Landi, D.; Cecchi, E.; Chiostri, M.; Gensini, G.F.; Valente, S.; Ciaccheri, M.; Castelli, G.; Romano, S.M. Effects of ULTRAfiltration vs. DIureticS on Clinical, Biohumoral and Haemodynamic Variables in Patients with DeCOmpensated Heart Failure: The ULTRADISCO Study. Eur. J. Heart. Fail. 2011, 13, 337–346. [Google Scholar] [CrossRef]
- Costanzo, M.R.; Guglin, M.E.; Saltzberg, M.T.; Jessup, M.L.; Bart, B.A.; Teerlink, J.R.; Jaski, B.E.; Fang, J.C.; Feller, E.D.; Haas, G.J.; et al. Ultrafiltration versus Intravenous Diuretics for Patients Hospitalized for Acute Decompensated Heart Failure. J. Am. Coll. Cardiol. 2007, 49, 675–683. [Google Scholar] [CrossRef]
- Bart, B.A.; Boyle, A.; Bank, A.J.; Anand, I.; Olivari, M.T.; Kraemer, M.; Mackedanz, S.; Sobotka, P.A.; Schollmeyer, M.; Goldsmith, S.R. Ultrafiltration versus Usual Care for Hospitalized Patients with Heart Failure: The Relief for Acutely Fluid-Overloaded Patients with Decompensated Congestive Heart Failure (RAPID-CHF). Trial. J. Am. Coll. Cardiol. 2005, 46, 2043–2046. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signs and symptoms of ODS |
|
|
| First Author (Ref. #) | Year | Type of Study | Treatment | Effects of Hypertonic Saline Association to Loop Diuretics |
|---|---|---|---|---|
| Tuttolomondo et al. [53] | 2021 | Randomized Controlled Trial 141 patients | 30 min of i.v. infusion of furosemide (120–250 mg) + HSS (150 mL of 1.4–4.6% NaCl) twice a day for 6 days versus 30 min of i.v. infusion of furosemide (120–150 mg) twice a day without HSS for 6 days. | Increase in diuresis, weight loss; reduction in the serum markers of atrial stretching, fibrosis and inflammation. |
| Griffin et al. [54] | 2020 | Retrospective Study 40 patients | i.v. 150 mL of 3% NaCl over 30 min + high doses of loop diuretics. | Improvements in urine output, weight loss, diuretic efficiency, renal function; increase in serum sodium levels; no discernible deterioration in respiratory status or overcorrection of hyponatremia. |
| Wan et al. [55] | 2017 | Randomized Controlled Trial 264 patients | i.v. 1-h infusion of furosemide (100 mg) plus HSS (100 mL) twice a day and severe water restriction (< 500 mL) vs. i.v. furosemide (100 mg) twice a day and severe water restriction (< 500 mL) without HSS. | Increase in urination, reduction of hospitalization time and costs; higher average readmission time; lower mortality rate. |
| Lafrenière et al. [56] | 2017 | Prospective Study 47 patients | i.v. infusion of 250 mg furosemide plus 150 mL HSS 3% NaCl twice a day for a mean duration of 2.3 days. | Greater weight loss per day of treatment. |
| Yayla et al. [57] | 2015 | Randomized Controlled Trial 43 patients | Continuous infusion of 160 mg furosemide for 16 h/day versus bolus injections of 80 mg furosemide twice a day versus administration of 160 mg furosemide plus HSS as an infusion for 30 min once a day. | Significantly shorter hospitalization. |
| Paterna et al. [58] | 2015 | Randomized Controlled Trial 40 patients | i.v. furosemide (125 mg/250 mg/500 mg) diluted in 150 mL of normal saline. (0.9%) versus the same furosemide dose diluted in 150 mL of HSS (1.4%). | Increased total urine output, sodium excretion, urinary osmolality, and furosemide urine delivery. |
| Okuhara et al. [59] | 2014 | Randomized Controlled Trial 44 patients | 1.7% HSS with 40 mg furosemide versus glucose 5% with 40 mg furosemide. | Favorable diuresis through increasing glomerular filtration rate. |
| Issa et al. [60] | 2013 | Randomized Controlled Trial 34 patients | Three-day course of 100 mL HSS (NaCl 7.5%) twice a day versus placebo. | Improvement in glomerular and tubular defects; attenuation of heart failure-induced kidney disfunction. |
| Parrinello et al. [61] | 2012 | Randomized Controlled Trial 248 patients | i.v. 30 min infusion of 250 mg furosemide twice a day with versus without HSS (1.4–4.6% NaCl). | Significant improvement in renal function, hydration state, pulmonary capillary wedge pressure, end diastolic volume, ejection fraction; increase in serum sodium levels; significant reduction in body weight, cardiac troponin I and brain natriuretic peptide, and hospitalization time. |
| Parrinello et al. [62] | 2011 | Randomized Controlled Trial 133 patients | i.v. infusion of 250 mg furosemide plus 150 mL 3% hypertonic saline twice a day and light sodium restriction (120 mmol) versus i.v. infusion of 250 mg furosemide twice a day and low sodium diet (80 mmol). | Significant improvement in renal function; increase in diuresis, natriuresis and serum sodium levels; faster reduction of echocardiographic pulmonary capillary wedge pressure. |
| Paterna et al. [63] | 2011 | Randomized Controlled Trial 1771 patients | i.v. infusion of 250 mg furosemide plus 150 mL 3% hypertonic saline twice a day and light sodium restriction (120 mmol) versus i.v. infusion of 250 mg furosemide twice a day and low sodium diet (80 mmol). | Increase in diuresis, natriuresis and serum sodium levels; reduction of hospitalization time; lower rate of readmission; lower rate of mortality. |
| Paterna et al. [64] | 2005 | Randomized Controlled Trial 94 patients | i.v. 500–1000 mg furosemide with 150 mL HSS (1.4–4.6% NaCl) twice a day versus i.v. 500-1000 mg furosemide twice a day without HSS. | Increase in diuresis, natriuresis and serum sodium levels; decrease in brain natriuretic peptide levels; reduction in hospitalization time and readmission rate. |
| Licata et al. [65] | 2003 | Randomized Controlled Trial 107 patients | i.v. 30 min infusion of 500–1000 mg furosemide with 150 mL HSS (1.4–4.6% NaCl) twice a day versus i.v. 500–1000 mg furosemide twice a day without HSS. | Significant increase in diuresis and natriuresis; increase in serum sodium levels; mortality reduction. |
| Paterna et al. [66] | 2000 | Randomized Controlled Trial 60 patients | i.v. 500–1000 mg furosemide with 150 mL HSS (1.4–4.6% NaCl) twice a day versus i.v. 500–1000 mg furosemide twice a day without HSS. | Increase in diuresis, natriuresis, and serum sodium levels; decrease in serum creatinine; reduction of hospitalization time; maintaining the obtained results over time. |
| Udelson 2011 [74] | AQUA-AHF 2020 [75] | |
|---|---|---|
| Enrolled patients | 83 Multicenter, randomized, double-blind placebo-controlled. | 33 Prospective, randomized, open-label, parallel-group, single center study. |
| Inclusion criteria | NYHA class II or III HF, systolic dysfunction (EF ≤ 40%) and signs of congestion (edema, rales). | Acute congestive HF and a serum Na < 135 mmol/L. |
| Design/tolvaptan dosing | 4 groups: tolvaptan 30 mg, furosemide 80 mg, a combination of tolvaptan 30 mg/furosemide 80 mg and placebo for 7 days (+ standard therapy). | Randomized to receive tolvaptan 30 mg orally daily or furosemide 5 mg/h intravenously for 24 h, after which treatment could be escalated. |
| Primary end point results | Reduction of body weight was similar in all groups. | No significant differences in median urine output or net fluid balance between groups at 24 h. |
| Secondary end point results | An increase in serum sodium within the normal range was also observed in tolvaptan-treated group when compared with placebo or furosemide group. | Oral tolvaptan was associated with similar, but not superior diuresis compared with intravenous furosemide for acute HF with concomitant hyponatremia. In contrast to conventional diuretics, exacerbation of hyponatremia is unlikely with tolvaptan. |
| EVEREST 2007 | TACTICS 2017 | SECRET of CHF 2017 | ACTIV in CHF 2004 | SALT 1 and SALT 2 2006 | |
|---|---|---|---|---|---|
| Enrolled patients | 4133 Phase III clinical trial | 257 Multicenter, randomized double-blind, placebo controlled trial | 250 Randomized, double-blind, placebo-controlled trial | 319 Prospective, international, multicenter, randomized controlled trial | 448 Multicenter, randomized, double-blind, placebo controlled trial |
| Enrollment time after admission | Within 48 h | Within 24 h | Within 36 h | Within 72 h | |
| Inclusion criteria | NYHA class III or IV ≥ 2 signs/symptoms of fluid overload (dyspnea, peripheral edema, and JVD) EF ≤ 40%. | Dyspnea at rest, BNP > 400 pg/mL, NT-proBNP > 2000 pg/mL, orthopnea or peripheral edema or JVD or pulmonary rales or congestion on chest X-ray, Serum sodium ≤ 140 mmol/L. | NYHA class III or IV or acute decompensated HF (dyspnea and either impaired renal function, diuretic resistance or hyponatremia). | Decompensated HF, EF < 40%, fluid overload requiring hospitalization. | Hyponatremia arising from CHF, cirrhosis and SIADH. |
| Exclusion criteria | SBP < 90 mmHg, Serum creatinine > 3.5 mg/dL or dialysis, Serum potassium > 5.5 mEq/L. | Serum sodium > 140 mEq/L, SBP < 90 mmHg, Serum creatinine> 3.5 mg/dL or currently undergoing renal replacement therapy. | SBP < 90 mmHg, Serum sodium > 144 mEq/L, Serum creatinine > 3.5 mg/dL or dialysis. | Recent myocardial infarction, recent cardiac surgery, SBP < 110 mmHg, life-threatening arrhythmias, severe renal impairment. | |
| Tolvaptan dosing | 30 mg of tolvaptan in addition of standard therapy for a minimum of 60 days. | Tolvaptan 30 mg orally vs. placebo given at 0, 24, and 48 h (e.g., 3 doses) in patients hospitalized for AHF and congestion in addition to a fixed dose of furosemide. | 30 mg of tolvaptan vs placebo during hospitalization (for up to 7 days). | Tolvaptan orally (30, 60 or 90 mg/day) or placebo (randomized 1:1:1:1) + standard therapy, including diuretics. Study drug continued for 60 days. | Tolvaptan 15 mg with titration to 30 and 60 mg as needed to correct sodium over 30 days, with a follow-up visit 7 days after study end. |
| Primary end point results | Greater reduction in body weight at day 1 and day 7 (or at time of discharge). | No difference in the percentage of patients who showed moderate or marked clinical improvement with tolvaptan (without need for rescue therapy). | No difference in self –assessment of dyspnea at 8 and 16 h. | Greater urine output and weight loss at 24 h after initial dose of tolvaptan when compared to the placebo group. | Serum sodium concentration increased more in tolvaptan group during the first 4 days and after the full 30 days of therapy. Within 7 days after stopping tolvaptan, serum sodium returned to the placebo levels. |
| Secondary end point results | Improved dyspnea and peripheral edema. No excess of renal failure or hypotension in the tolvaptan group. Tolvaptan significantly increased serum sodium levels at day 7. | More weight loss and urine output at 24 h with tolvaptan. No difference in dyspnea relief. Serum sodium increased by an average of 3 mmol/L in the tolvaptan group. | Greater weight reduction with tolvaptan. By day 3 there was a greater reduction in dyspnea in the tolvaptan group. Hyponatremia as an adverse event in tolvaptan group was reported, but without statistical significance compared to placebo. | Less dyspnea at the time of discharge. No difference in progression of HF. Tolvaptan use reduced the amount of loop diuretic used. No difference in HR, BP, or renal function. Tolvaptan was associated with normalization of serum sodium in patients with hyponatremia. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Şorodoc, V.; Asaftei, A.; Puha, G.; Ceasovschih, A.; Lionte, C.; Sîrbu, O.; Bologa, C.; Haliga, R.E.; Constantin, M.; Coman, A.E.; et al. Management of Hyponatremia in Heart Failure: Practical Considerations. J. Pers. Med. 2023, 13, 140. https://doi.org/10.3390/jpm13010140
Şorodoc V, Asaftei A, Puha G, Ceasovschih A, Lionte C, Sîrbu O, Bologa C, Haliga RE, Constantin M, Coman AE, et al. Management of Hyponatremia in Heart Failure: Practical Considerations. Journal of Personalized Medicine. 2023; 13(1):140. https://doi.org/10.3390/jpm13010140
Chicago/Turabian StyleŞorodoc, Victoriţa, Andreea Asaftei, Gabriela Puha, Alexandr Ceasovschih, Cătălina Lionte, Oana Sîrbu, Cristina Bologa, Raluca Ecaterina Haliga, Mihai Constantin, Adorata Elena Coman, and et al. 2023. "Management of Hyponatremia in Heart Failure: Practical Considerations" Journal of Personalized Medicine 13, no. 1: 140. https://doi.org/10.3390/jpm13010140