
Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis



Abstract
:1. Introduction
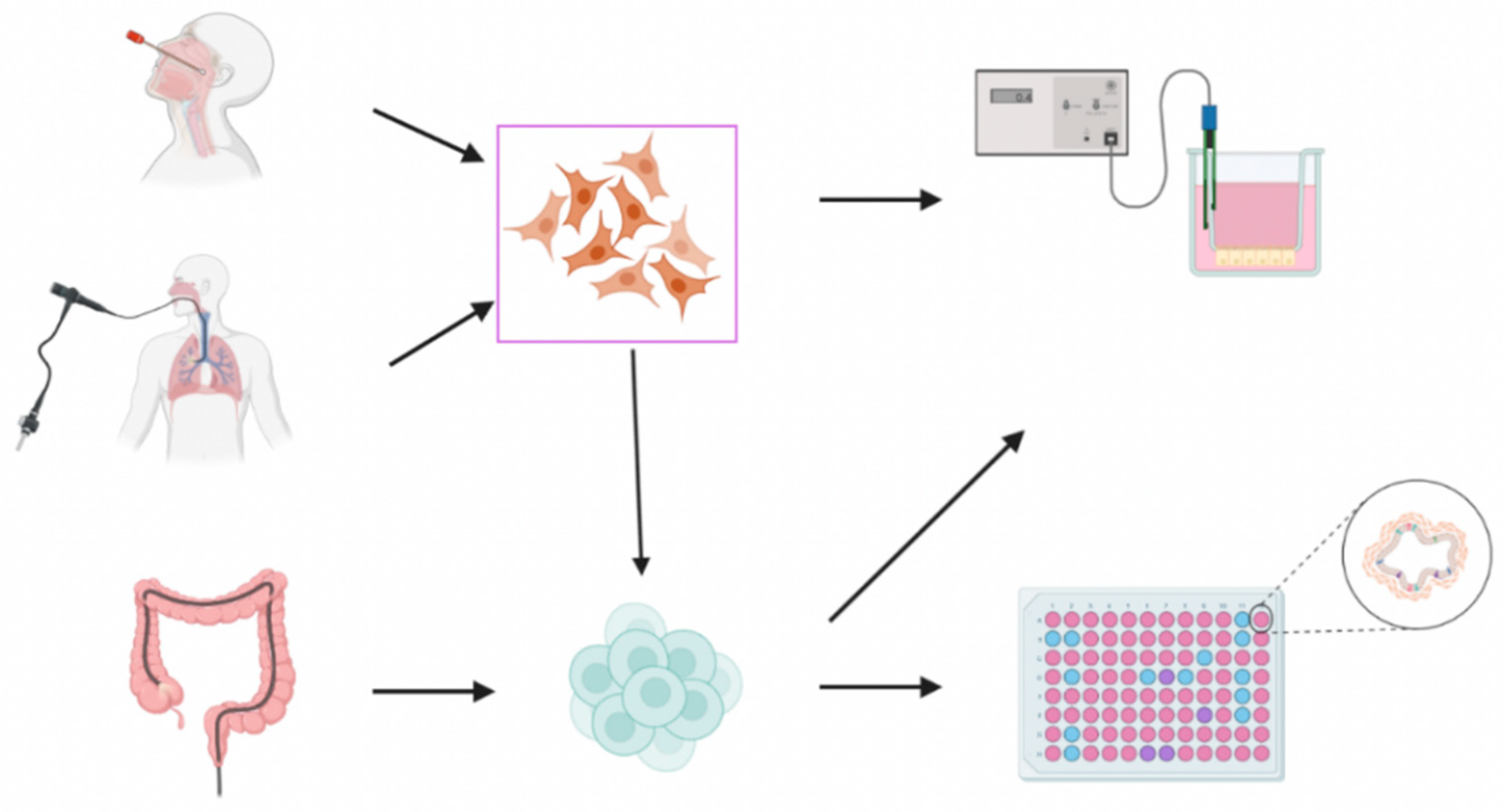
2. Preclinical In Vitro Models
2.1. Cell Lines
2.2. Patient-Derived Samples
2.2.1. Lung/Airway Models
- Primary Airway Cells in Planar Cultures
- Airway Organoids/Spheroids
2.2.2. Gastrointestinal Models
- Rectal Biopsies
- Intestinal Organoids
- Intestinal Organoids-Derived 2D Monolayers
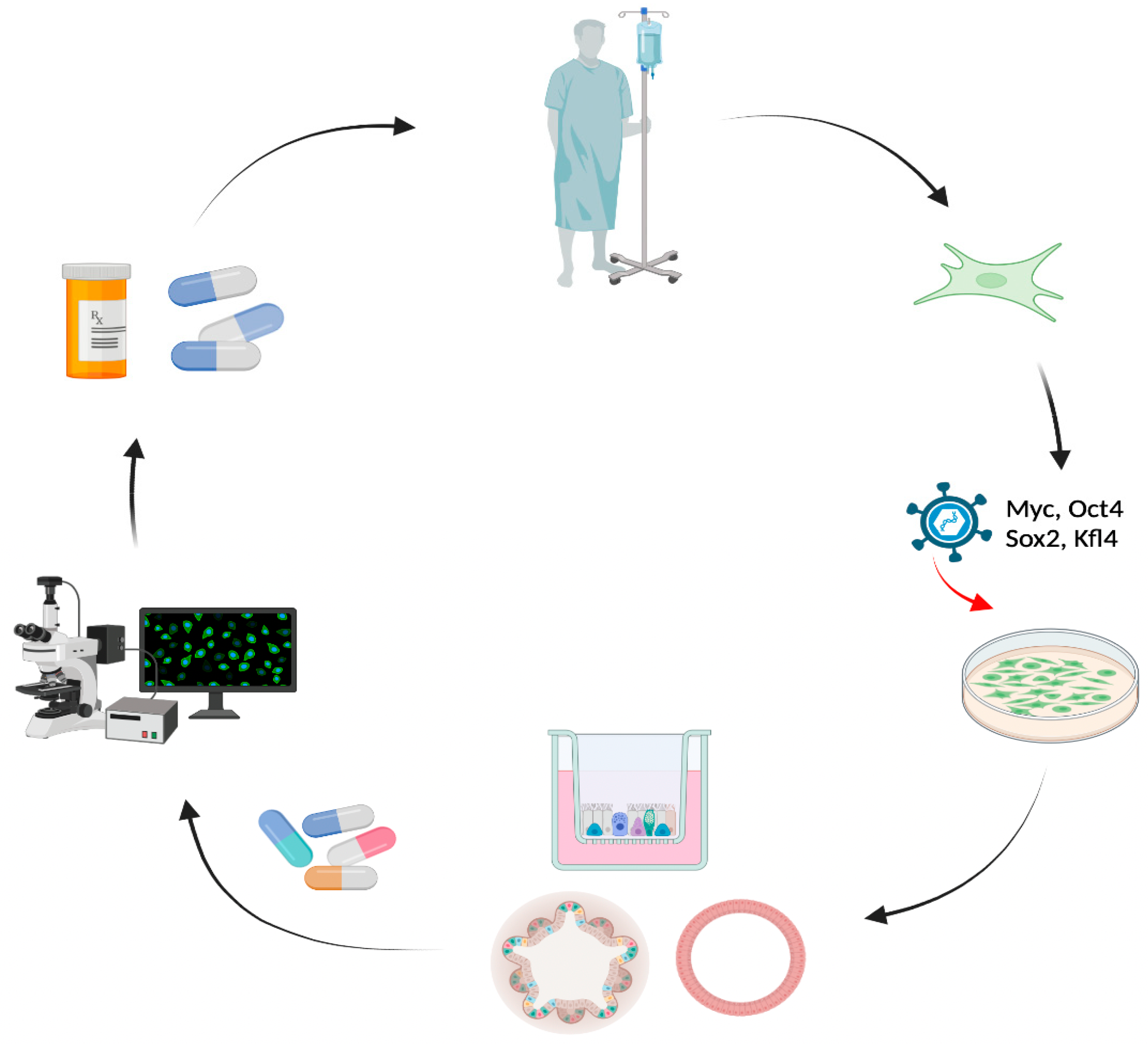
2.3. Induced Pluripotent Stem Cells (iPSCs)
2.4. Organ-on-a-Chip System
3. Outlook and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Prim. 2015, 1, 15010. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.A.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front. Pharmacol. 2016, 7, 275. [Google Scholar] [CrossRef]
- Cohen-Cymberknoh, M.; Shoseyov, D.; Kerem, E. Managing Cystic Fibrosis: Strategies that increase life expectancy and improve quality of life. Am. J. Respir. Crit. Care Med. 2011, 183, 1463–1471. [Google Scholar] [CrossRef]
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Pedemonte, N.; Kicic, A. Editorial: Emerging Therapeutic Approaches for Cystic Fibrosis. Front. Pharmacol. 2019, 10, 1440. [Google Scholar] [CrossRef]
- Narayanan, S.; Mainz, J.G.; Gala, S.; Tabori, H.; Grossoehme, D. Adherence to therapies in cystic fibrosis: A targeted literature review. Expert Rev. Respir. Med. 2017, 11, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.C.; Silva, I.A.L.; Figueira, M.F.; Amaral, M.D.; Lopes-Pacheco, M. Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J. Exp. Pharmacol. 2021, 13, 693–723. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin. Drug Discov. 2021, 16, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Gruenert, D.C.; Willems, M.; Cassiman, J.J.; Frizzell, R.A. Established cell lines used in cystic fibrosis research. J. Cyst. Fibros. 2004, 3, 191–196. [Google Scholar] [CrossRef]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef]
- Pedemonte, N.; Lukacs, G.L.; Du, K.; Caci, E.; Zegarra-Moran, O.; Galietta, L.J.V.; Verkman, A.S. Small-molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Investig. 2005, 115, 2564–2571. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Straley, K.S.; Cao, D.; González, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.; et al. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L1117–L1130. [Google Scholar] [CrossRef]
- Robert, R.; Carlile, G.W.; Pavel, C.; Liu, N.; Anjos, S.M.; Liao, J.; Luo, Y.; Zhang, D.; Thomas, D.Y.; Hanrahan, J.W. Structural Analog of Sildenafil Identified as a Novel Corrector of the F508del-CFTR Trafficking Defect. Mol. Pharmacol. 2008, 73, 478–489. [Google Scholar] [CrossRef]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Davies, J.C.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef]
- McKone, E.F.; Borowitz, D.; Drevinek, P.; Griese, M.; Konstan, M.W.; Wainwright, C.; Ratjen, F.; Sermet-Gaudelus, I.; Plant, B.; Munck, A.; et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp- CFTR mutation: A phase 3, open-label extension study (PERSIST). Lancet Respir. Med. 2014, 2, 902–910. [Google Scholar] [CrossRef]
- Clancy, J.P.; Rowe, S.M.; Accurso, F.J.; Aitken, M.L.; Amin, R.S.; Ashlock, M.A.; Ballmann, M.; Boyle, M.P.; Bronsveld, I.; Campbell, P.W.; et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for theF508del-CFTR mutation. Thorax 2012, 67, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Flume, P.A.; Liou, T.G.; Borowitz, D.S.; Li, H.; Yen, K.; Ordoñez, C.L.; Geller, D.E. Ivacaftor in Subjects with Cystic Fibrosis Who Are Homozygous for the F508del-CFTR Mutation. Chest 2012, 142, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; McKone, E.F.; Moss, R.B.; Marigowda, G.; Tian, S.; Waltz, D.; Huang, X.; Lubarsky, B.; Rubin, J.; Millar, S.J.; et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): A phase 3, extension study. Lancet Respir. Med. 2017, 5, 107–118. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Costa, E.; Girotti, S.; Pauro, F.; Leufkens, H.G.M.; Cipolli, M. The impact of FDA and EMA regulatory decision-making process on the access to CFTR modulators for the treatment of cystic fibrosis. Orphanet J. Rare Dis. 2022, 17, 188. [Google Scholar] [CrossRef]
- Farinha, C.M.; Sousa, M.; Canato, S.; Schmidt, A.; Uliyakina, I.; Amaral, M.D. Increased efficacy of VX -809 in different cellular systems results from an early stabilization effect of F508del-CFTR. Pharmacol. Res. Perspect. 2015, 3, e00152. [Google Scholar] [CrossRef] [PubMed]
- Avramescu, R.G.; Kai, Y.; Xu, H.; Bidaud-Meynard, A.; Schnúr, A.; Frenkiel, S.; Matouk, E.; Veit, G.; Lukacs, G.L. Mutation-specific downregulation of CFTR2 variants by gating potentiators. Hum. Mol. Genet. 2017, 26, 4873–4885. [Google Scholar] [CrossRef] [PubMed]
- Mogayzel, P.J.; Henning, K.A.; Bittner, M.L.; Novotny, E.A.; Schwiebert, E.M.; Guggino, W.B.; Jiang, Y.; Rosenfeld, M.A. Functional human CFTR produced by stable Chinese hamster ovary cell lines derived using yeast artificial chromosomes. Hum. Mol. Genet. 1997, 6, 59–68. [Google Scholar] [CrossRef]
- Weber, A.J.; Soong, G.; Bryan, R.; Saba, S.; Prince, A. Activation of NF-κB in airway epithelial cells is dependent on CFTR trafficking and Cl− channel function. Am. J. Physiol. Cell. Mol. Physiol. 2001, 281, L71–L78. [Google Scholar] [CrossRef]
- Bose, S.J.; Bijvelds, M.J.C.; Wang, Y.; Liu, J.; Cai, Z.; Bot, A.G.M.; de Jonge, H.R.; Sheppard, D.N. Differential thermostability and response to cystic fibrosis transmembrane conductance regulator potentiators of human and mouse F508del-CFTR. Am. J. Physiol. Cell. Mol. Physiol. 2019, 317, L71–L86. [Google Scholar] [CrossRef] [PubMed]
- Seibert, F.S.; Linsdell, P.; Loo, T.W.; Hanrahan, J.W.; Riordan, J.R.; Clarke, D.M. Cytoplasmic Loop Three of Cystic Fibrosis Transmembrane Conductance Regulator Contributes to Regulation of Chloride Channel Activity. J. Biol. Chem. 1996, 271, 27493–27499. [Google Scholar] [CrossRef]
- Rapino, D.; Sabirzhanova, I.; Lopes-Pacheco, M.; Grover, R.; Guggino, W.B.; Cebotaru, L. Rescue of NBD2 Mutants N1303K and S1235R of CFTR by Small-Molecule Correctors and Transcomplementation. PLoS ONE 2015, 10, e0119796. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Sabirzhanova, I.; Rapino, D.; Morales, M.M.; Guggino, W.B.; Cebotaru, L. Correctors Rescue CFTR Mutations in Nucleotide-Binding Domain 1 (NBD1) by Modulating Proteostasis. ChemBioChem 2016, 17, 493–505. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Boinot, C.; Sabirzhanova, I.; Rapino, D.; Cebotaru, L.L. Combination of Correctors Rescues CFTR Transmembrane-Domain Mutants by Mitigating their Interactions with Proteostasis. Cell. Physiol. Biochem. 2017, 41, 2194–2210. [Google Scholar] [CrossRef]
- Caputo, A.; Hinzpeter, A.; Caci, E.; Pedemonte, N.; Arous, N.; Di Duca, M.; Zegarra-Moran, O.; Fanen, P.; Galietta, L.J.V. Mutation-Specific Potency and Efficacy of Cystic Fibrosis Transmembrane Conductance Regulator Chloride Channel Potentiators. J. Pharmacol. Exp. Ther. 2009, 330, 786–791. [Google Scholar] [CrossRef]
- Lee, M.G.; Wigley, W.C.; Zeng, W.; Noel, L.E.; Marino, C.R.; Thomas, P.J.; Muallem, S. Regulation of Cl−/HCO3−Exchange by Cystic Fibrosis Transmembrane Conductance Regulator Expressed in NIH 3T3 and HEK 293 Cells. J. Biol. Chem. 1999, 274, 3414–3421. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M.; Boinot, C.; Sabirzhanova, I.; Morales, M.M.; Guggino, W.B.; Cebotaru, L. Combination of Correctors Rescue ΔF508-CFTR by Reducing Its Association with Hsp40 and Hsp27. J. Biol. Chem. 2015, 290, 25636–25645. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Evans, T.A.; Pellicore, M.J.; Davis, E.; Aksit, M.A.; McCague, A.F.; Joynt, A.T.; Lu, Z.; Han, S.T.; Anzmann, A.F.; et al. Capitalizing on the heterogeneous effects of CFTR nonsense and frameshift variants to inform therapeutic strategy for cystic fibrosis. PLoS Genet. 2018, 14, e1007723. [Google Scholar] [CrossRef] [PubMed]
- Patel, W.; Moore, P.J.; Sassano, M.F.; Lopes-Pacheco, M.; Aleksandrov, A.A.; Amaral, M.D.; Tarran, R.; Gray, M.A. Increases in cytosolic Ca2+ induce dynamin- and calcineurin-dependent internalisation of CFTR. Cell. Mol. Life Sci. 2019, 76, 977–994. [Google Scholar] [CrossRef] [PubMed]
- Moyer, B.D.; Loffing, J.; Schwiebert, E.M.; Loffing-Cueni, D.; Halpin, P.A.; Karlson, K.H.; Ismailov, I.I.; Guggino, W.B.; Langford, G.M.; Stanton, B.A. Membrane Trafficking of the Cystic Fibrosis Gene Product, Cystic Fibrosis Transmembrane Conductance Regulator, Tagged with Green Fluorescent Protein in Madin-Darby Canine Kidney Cells. J. Biol. Chem. 1998, 273, 21759–21768. [Google Scholar] [CrossRef]
- Krasnov, K.V.; Tzetis, M.; Cheng, J.; Guggino, W.B.; Cutting, G.R. Localization studies of rare missense mutations in cystic fibrosis transmembrane conductance regulator (CFTR) facilitate interpretation of genotype-phenotype relationships. Hum. Mutat. 2008, 29, 1364–1372. [Google Scholar] [CrossRef]
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 2014, 13, 29–36. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Silva, I.A.L.; Turner, M.J.; Carlile, G.W.; Sondo, E.; Thomas, D.Y.; Pedemonte, N.; Hanrahan, J.W.; Amaral, M.D. Characterization of the mechanism of action of RDR01752, a novel corrector of F508del-CFTR. Biochem. Pharmacol. 2020, 180, 114133. [Google Scholar] [CrossRef]
- Han, S.T.; Rab, A.; Pellicore, M.J.; Davis, E.F.; McCague, A.F.; Evans, T.A.; Joynt, A.T.; Lu, Z.; Cai, Z.; Raraigh, K.S.; et al. Residual function of cystic fibrosis mutants predicts response to small molecule CFTR modulators. JCI Insight 2018, 3, e121159. [Google Scholar] [CrossRef]
- Cozens, A.L.; Yezzi, M.J.; Kunzelmann, K.; Ohrui, T.; Chin, L.; Eng, K.; Finkbeiner, W.E.; Widdicombe, J.H.; Gruenert, D.C. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1994, 10, 38–47. [Google Scholar] [CrossRef]
- Bebok, Z.; Collawn, J.F.; Wakefield, J.; Parker, W.; Li, Y.; Varga, K.; Sorscher, E.J.; Clancy, J.P. Failure of cAMP agonists to activate rescued ΔF508 CFTR in CFBE41o− airway epithelial monolayers. J. Physiol. 2005, 569, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M.; Bacalhau, M.; Ramalho, S.S.; Silva, I.A.L.; Ferreira, F.C.; Carlile, G.W.; Thomas, D.Y.; Farinha, C.M.; Hanrahan, J.W.; Amaral, M.D. Rescue of Mutant CFTR Trafficking Defect by the Investigational Compound MCG1516A. Cells 2022, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Burton, B.; Huang, C.J.; Worley, J.; Cao, D.; Johnson, J.P.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012, 11, 237–245. [Google Scholar] [CrossRef]
- Amato, F.; Scudieri, P.; Musante, I.; Tomati, V.; Caci, E.; Comegna, M.; Maietta, S.; Manzoni, F.; Di Lullo, A.M.; De Wachter, E.; et al. Two CFTR mutations within codon 970 differently impact on the chloride channel functionality. Hum. Mutat. 2019, 40, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Fidler, M.C.; Buckley, A.; Sullivan, J.C.; Statia, M.; Boj, S.F.; Vries, R.G.J.; Munck, A.; Higgins, M.; Moretto Zita, M.; Negulescu, P.; et al. G970R-CFTR Mutation (c.2908G>C) Results Predominantly in a Splicing Defect. Clin. Transl. Sci. 2021, 14, 656–663. [Google Scholar] [CrossRef]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef]
- Oren, Y.S.; Irony-Tur Sinai, M.; Golec, A.; Barchad-Avitzur, O.; Mutyam, V.; Li, Y.; Hong, J.; Ozeri-Galai, E.; Hatton, A.; Leibson, C.; et al. Antisense oligonucleotide-based drug development for Cystic Fibrosis patients carrying the 3849 + 10 kb C-to-T splicing mutation. J. Cyst. Fibros. 2021, 20, 865–875. [Google Scholar] [CrossRef]
- Nissim-Rafinia, M.; Aviram, M.; Randell, S.H.; Shushi, L.; Ozeri, E.; Chiba-Falek, O.; Eidelman, O.; Pollard, H.B.; Yankaskas, J.R.; Kerem, B. Restoration of the cystic fibrosis transmembrane conductance regulator function by splicing modulation. EMBO Rep. 2004, 5, 1071–1077. [Google Scholar] [CrossRef]
- Joynt, A.T.; Evans, T.A.; Pellicore, M.J.; Davis-Marcisak, E.F.; Aksit, M.A.; Eastman, A.C.; Patel, S.U.; Paul, K.C.; Osorio, D.L.; Bowling, A.D.; et al. Evaluation of both exonic and intronic variants for effects on RNA splicing allows for accurate assessment of the effectiveness of precision therapies. PLoS Genet. 2020, 16, e1009100. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Collnot, E.M.; Baldes, C.; Becker, U.; Laue, M.; Kim, K.J.; Lehr, C.M. Towards an in vitro model of cystic fibrosis small airway epithelium: Characterisation of the human bronchial epithelial cell line CFBE41o−. Cell Tissue Res. 2006, 323, 405–415. [Google Scholar] [CrossRef]
- Lundberg, A.S.; Randell, S.H.; Stewart, S.A.; Elenbaas, B.; Hartwell, K.A.; Brooks, M.W.; Fleming, M.D.; Olsen, J.C.; Miller, S.W.; Weinberg, R.A.; et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene 2002, 21, 4577–4586. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Tomati, V.; Sondo, E.; Galietta, L.J.V. Influence of cell background on pharmacological rescue of mutant CFTR. Am. J. Physiol. Physiol. 2010, 298, C866–C874. [Google Scholar] [CrossRef] [PubMed]
- Sondo, E.; Tomati, V.; Caci, E.; Esposito, A.I.; Pfeffer, U.; Pedemonte, N.; Galietta, L.J.V. Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am. J. Physiol. Physiol. 2011, 301, C872–C885. [Google Scholar] [CrossRef] [PubMed]
- Ostedgaard, L.S.; Rogers, C.S.; Dong, Q.; Randak, C.O.; Vermeer, D.W.; Rokhlina, T.; Karp, P.H.; Welsh, M.J. Processing and function of CFTR-ΔF508 are species-dependent. Proc. Natl. Acad. Sci. USA 2007, 104, 15370–15375. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, L.B.; Vecchio-Pagan, B.; Sharma, N.; Han, S.T.; Franca, A.; Wohler, E.S.; Batista, D.A.S.; Goff, L.A.; Cutting, G.R. Creation and characterization of an airway epithelial cell line for stable expression of CFTR variants. J. Cyst. Fibros. 2015, 15, 285–294. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Kneuer, C.; Fiegel, J.; Hanes, J.; Schaefer, U.; Kim, K.J.; Lehr, C.M. Influence of apical fluid volume on the development of functional intercellular junctions in the human epithelial cell line 16HBE14o-: Implications for the use of this cell line as an in vitro model for bronchial drug absorption studies. Cell Tissue Res. 2002, 308, 391–400. [Google Scholar] [CrossRef]
- Valley, H.C.; Bukis, K.M.; Bell, A.; Cheng, Y.; Wong, E.; Jordan, N.J.; Allaire, N.E.; Sivachenko, A.; Liang, F.; Bihler, H.; et al. Isogenic cell models of cystic fibrosis-causing variants in natively expressing pulmonary epithelial cells. J. Cyst. Fibros. 2019, 18, 476–483. [Google Scholar] [CrossRef]
- Venturini, A.; Borrelli, A.; Musante, I.; Scudieri, P.; Capurro, V.; Renda, M.; Pedemonte, N.; Galietta, L.J.V. Comprehensive Analysis of Combinatorial Pharmacological Treatments to Correct Nonsense Mutations in the CFTR Gene. Int. J. Mol. Sci. 2021, 22, 11972. [Google Scholar] [CrossRef]
- Laselva, O.; Bartlett, C.; Popa, A.; Ouyang, H.; Gunawardena, T.N.A.; Gonska, T.; Moraes, T.J.; Bear, C.E. Emerging preclinical modulators developed for F508del-CFTR have the potential to be effective for ORKAMBI resistant processing mutants. J. Cyst. Fibros. 2021, 20, 106–119. [Google Scholar] [CrossRef]
- Laselva, O.; McCormack, J.; Bartlett, C.; Ip, W.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Gonska, T.; Moraes, T.J.; Bear, C.E. Preclinical Studies of a Rare CFCausing Mutation in the Second Nucleotide Binding Domain (c.3700A>G) Show Robust Functional Rescue in Primary Nasal Cultures by Novel CFTR Modulators. J. Pers. Med. 2020, 10, 209. [Google Scholar] [CrossRef]
- Phuan, P.W.; Haggie, P.M.; Tan, J.A.; Rivera, A.A.; Finkbeiner, W.E.; Nielson, D.W.; Thomas, M.M.; Janahi, I.A.; Verkman, A.S. CFTR modulator therapy for cystic fibrosis caused by the rare c.3700A>G mutation. J. Cyst. Fibros. 2021, 20, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Mention, K.; Cavusoglu-Doran, K.; Sanz, D.J.; Bacalhau, M.; Lopes-Pacheco, M.; Harrison, P.T.; Farinha, C.M. Comparison of Cas9 and Cas12a CRISPR editing methods to correct the W1282XCFTR mutation. J. Cyst. Fibros. 2022, 21, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Henderson, J.M.; Coote, K.; Cheng, Y.; Valley, H.C.; Zhang, X.O.; Wang, Q.; Rhym, L.H.; Cao, Y.; Newby, G.A.; et al. Chemical modifications of adenine base editor mRNA and guide RNA expand its application scope. Nat. Commun. 2020, 11, 1979. [Google Scholar] [CrossRef] [PubMed]
- Guimbellot, J.; Solomon, G.M.; Baines, A.; Heltshe, S.L.; VanDalfsen, J.; Joseloff, E.; Sagel, S.D.; Rowe, S.M. Effectiveness of ivacaftor in cystic fibrosis patients with non-G551D gating mutations. J. Cyst. Fibros. 2019, 18, 102–109. [Google Scholar] [CrossRef]
- Salvatore, D.; Carnovale, V.; Iacotucci, P.; Braggion, C.; Castellani, C.; Cimino, G.; Colangelo, C.; Francalanci, M.; Leonetti, G.; Lucidi, V.; et al. Effectivenesss of ivacaftor in severe cystic fibrosis patients and non-G551D gating mutations. Pediatr. Pulmonol. 2019, 54, 1398–1403. [Google Scholar] [CrossRef]
- McCague, A.F.; Raraigh, K.S.; Pellicore, M.J.; Davis-Marcisak, E.F.; Evans, T.A.; Han, S.T.; Lu, Z.; Joynt, A.T.; Sharma, N.; Castellani, C.; et al. Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1116–1126. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; de Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; van de Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708. [Google Scholar] [CrossRef]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 375. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Pilewski, J.M.; Griese, M.; Cooke, J.; Viswanathan, L.; Tullis, E.; Davies, J.C.; Lekstrom-Himes, J.A.; Wang, L.T. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551DCFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. [Google Scholar] [CrossRef]
- Sepahzad, A.; Morris-Rosendahl, D.J.; Davies, J.C. Cystic Fibrosis Lung Disease Modifiers and Their Relevance in the New Era of Precision Medicine. Genes 2021, 12, 562. [Google Scholar] [CrossRef] [PubMed]
- McGarry, M.E.; McColley, S.A. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr. Pulmonol. 2021, 56, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef]
- Fulcher, M.L.; Randell, S.H. Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol. Biol. 2013, 945, 109–121. [Google Scholar] [CrossRef]
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK Inhibitor and Feeder Cells Induce the Conditional Reprogramming of Epithelial Cells. Am. J. Pathol. 2012, 180, 599–607. [Google Scholar] [CrossRef]
- Liu, X.; Krawczyk, E.; Suprynowicz, F.A.; Palechor-Ceron, N.; Yuan, H.; Dakic, A.; Simic, V.; Zheng, Y.L.; Sripadhan, P.; Chen, C.; et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat. Protoc. 2017, 12, 439–451. [Google Scholar] [CrossRef]
- Mou, H.; Vinarsky, V.; Tata, P.R.; Brazauskas, K.; Choi, S.H.; Crooke, A.K.; Zhang, B.; Solomon, G.M.; Turner, B.; Bihler, H.; et al. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell Stem Cell 2016, 19, 217–231. [Google Scholar] [CrossRef]
- Gentzsch, M.; Boyles, S.E.; Cheluvaraju, C.; Chaudhry, I.G.; Quinney, N.L.; Cho, C.; Dang, H.; Liu, X.; Schlegel, R.; Randell, S.H. Pharmacological Rescue of Conditionally Reprogrammed Cystic Fibrosis Bronchial Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2017, 56, 568–574. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S.; Li, M.; Li, J.; Shen, J.; Zhao, Y.; Pang, J.; Wen, Q.; Chen, M.; Wei, B.; et al. Conditional reprogramming: Next generation cell culture. Acta Pharm. Sin. B 2020, 10, 1360–1381. [Google Scholar] [CrossRef]
- Itani, O.A.; Chen, J.H.; Karp, P.H.; Ernst, S.; Keshavjee, S.; Parekh, K.; Klesney-Tait, J.; Zabner, J.; Welsh, M.J. Human cystic fibrosis airway epithelia have reduced Cl− conductance but not increased Na+ conductance. Proc. Natl. Acad. Sci. USA 2011, 108, 10260–10265. [Google Scholar] [CrossRef] [PubMed]
- Coakley, R.D.; Grubb, B.R.; Paradiso, A.M.; Gatzy, J.T.; Johnson, L.G.; Kreda, S.M.; O’Neal, W.K.; Boucher, R.C. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc. Natl. Acad. Sci. USA 2003, 100, 16083–16088. [Google Scholar] [CrossRef] [PubMed]
- Birket, S.E.; Chu, K.K.; Liu, L.; Houser, G.H.; Diephuis, B.J.; Wilsterman, E.J.; Dierksen, G.; Mazur, M.; Shastry, S.; Li, Y.; et al. A Functional Anatomic Defect of the Cystic Fibrosis Airway. Am. J. Respir. Crit. Care Med. 2014, 190, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.S.; Jiang, J.; Ahmadi, S.; Lew, A.; Laselva, O.; Xia, S.; Bartlett, C.; Ip, W.; Wellhauser, L.; Ouyang, H.; et al. ORKAMBIMediated Rescue of Mucociliary Clearance in Cystic Fibrosis Primary Respiratory Cultures Is Enhanced by Arginine Uptake, Arginase Inhibition, and Promotion of Nitric Oxide Signaling to the Cystic Fibrosis Transmembrane Conductance Regulator Channel. Mol. Pharmacol. 2019, 96, 515–525. [Google Scholar] [CrossRef]
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J.; Moncivaiz, J.D.; Ostmann, A.J.; Strecker, L.M.; Clancy, J.P. Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight 2018, 3, e99385. [Google Scholar] [CrossRef]
- Müller, L.; Brighton, L.E.; Carson, J.L.; Fischer, W.A.; Jaspers, I. Culturing of Human Nasal Epithelial Cells at the Air Liquid Interface. J. Vis. Exp. 2013, 80, e50646. [Google Scholar] [CrossRef]
- Sette, G.; Cicero, S.L.; Blaconà, G.; Pierandrei, S.; Bruno, S.M.; Salvati, V.; Castelli, G.; Falchi, M.; Fabrizzi, B.; Cimino, G.; et al. Theratyping cystic fibrosis in vitro in ALI culture and organoid models generated from patient-derived nasal epithelial conditionally reprogrammed stem cells. Eur. Respir. J. 2021, 58, 2100908. [Google Scholar] [CrossRef]
- Noel, S.; Servel, N.; Hatton, A.; Golec, A.; Rodrat, M.; Ng, D.R.S.; Li, H.; Pranke, I.; Hinzpeter, A.; Edelman, A.; et al. Correlating genotype with phenotype using CFTR-mediated whole-cell Cl − currents in human nasal epithelial cells. J. Physiol. 2022, 600, 1515–1531. [Google Scholar] [CrossRef]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef]
- Scudieri, P.; Musante, I.; Venturini, A.; Guidone, D.; Genovese, M.; Cresta, F.; Caci, E.; Palleschi, A.; Poeta, M.; Santamaria, F.; et al. Ionocytes and CFTR Chloride Channel Expression in Normal and Cystic Fibrosis Nasal and Bronchial Epithelial Cells. Cells 2020, 9, 2090. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Dang, H.; Kobayashi, Y.; Carraro, G.; Nakano, S.; Chen, G.; Kato, T.; Asakura, T.; Gilmore, R.C.; Morton, L.C.; et al. Secretory Cells Dominate Airway CFTR Expression and Function in Human Airway Superficial Epithelia. Am. J. Respir. Crit. Care Med. 2021, 203, 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Pranke, I.; Hatton, A.; Masson, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; Urbach, V.; Hinzpeter, A.; Edelman, A.; et al. Might Brushed Nasal Cells Be a Surrogate for CFTR Modulator Clinical Response? Am. J. Respir. Crit. Care Med. 2019, 199, 123–126. [Google Scholar] [CrossRef]
- McDougall, C.M.; Blaylock, M.G.; Douglas, J.G.; Brooker, R.J.; Helms, P.J.; Walsh, G.M. Nasal Epithelial Cells as Surrogates for Bronchial Epithelial Cells in Airway Inflammation Studies. Am. J. Respir. Cell Mol. Biol. 2008, 39, 560–568. [Google Scholar] [CrossRef] [PubMed]
- de Courcey, F.; Zholos, A.V.; Atherton-Watson, H.; Williams, M.T.S.; Canning, P.; Danahay, H.L.; Elborn, J.S.; Ennis, M. Development of primary human nasal epithelial cell cultures for the study of cystic fibrosis pathophysiology. Am. J. Physiol. Physiol. 2012, 303, C1173–C1179. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.D.; Liu, Z.; Odom, L.V.; Kersh, L.; Guimbellot, J.S. CFTR function and clinical response to modulators parallel nasal epithelial organoid swelling. Am. J. Physiol. Cell. Mol. Physiol. 2021, 321, L119–L129. [Google Scholar] [CrossRef]
- Zabner, J.; Couture, L.A.; Gregory, R.J.; Graham, S.M.; Smith, A.E.; Welsh, M. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell 1993, 75, 207–216. [Google Scholar] [CrossRef]
- Cao, H.; Ouyang, H.; Laselva, O.; Bartlett, C.; Zhou, Z.P.; Duan, C.; Gunawardena, T.; Avolio, J.; Bear, C.E.; Gonska, T.; et al. A helper-dependent adenoviral vector rescues CFTR to wild-type functional levels in cystic fibrosis epithelial cells harbouring class I mutations. Eur. Respir. J. 2020, 56, 2000205. [Google Scholar] [CrossRef]
- Oren, Y.S.; Avizur-Barchad, O.; Ozeri-Galai, E.; Elgrabli, R.; Schirelman, M.R.; Blinder, T.; Stampfer, C.D.; Ordan, M.; Laselva, O.; Cohen-Cymberknoh, M.; et al. Antisense oligonucleotide splicing modulation as a novel Cystic Fibrosis therapeutic approach for the W1282X nonsense mutation. J. Cyst. Fibros. 2021, 21, 630–636. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Kitoko, J.Z.; Morales, M.M.; Petrs-Silva, H.; Rocco, P.R.M. Self-complementary and tyrosine-mutant rAAV vectors enhance transduction in cystic fibrosis bronchial epithelial cells. Exp. Cell Res. 2018, 372, 99–107. [Google Scholar] [CrossRef]
- Roomans, G.M.; Kozlova, I.; Nilsson, H.; Vanthanouvong, V.; Button, B.; Tarran, R. Measurements of airway surface liquid height and mucus transport by fluorescence microscopy, and of ion composition by X-ray microanalysis. J. Cyst. Fibros. 2004, 3, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.Y.; Hwang, P.H.; Illek, B.; Fischer, H. Acid and base secretion in freshly excised nasal tissue from cystic fibrosis patients with ΔF508 mutation. Int. Forum Allergy Rhinol. 2011, 1, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Guimbellot, J.S.; Leach, J.M.; Chaudhry, I.G.; Quinney, N.L.; Boyles, S.E.; Chua, M.; Aban, I.; Jaspers, I.; Gentzsch, M. Nasospheroids permit measurements of CFTR-dependent fluid transport. JCI Insight 2017, 2, e95734. [Google Scholar] [CrossRef] [PubMed]
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J.; Ostmann, A.J.; Strecker, L.M.; Szczesniak, R.D.; Clancy, J.P. Detection of CFTR function and modulation in primary human nasal cell spheroids. J. Cyst. Fibros. 2018, 17, 26–33. [Google Scholar] [CrossRef]
- Debley, J.S.; Barrow, K.A.; Rich, L.M.; Singh, P.; McKone, E.F.; Nichols, D.P. Correlation between Ivacaftor-induced CFTR Activation in Airway Epithelial Cells and Improved Lung Function: A Proof-of-Concept Study. Ann. Am. Thorac. Soc. 2020, 17, 1024–1027. [Google Scholar] [CrossRef]
- McGarry, M.E.; Illek, B.; Ly, N.P.; Zlock, L.; Olshansky, S.; Moreno, C.; Finkbeiner, W.E.; Nielson, D.W. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: N-of-1 studies. Pediatr. Pulmonol. 2017, 52, 472–479. [Google Scholar] [CrossRef]
- Park, J.K.H.; Shrivastava, A.; Zhang, C.; Pollok, B.A.; Finkbeiner, W.E.; Gibb, E.R.; Ly, N.P.; Illek, B. Functional Profiling of CFTRDirected Therapeutics Using Pediatric Patient-Derived Nasal Epithelial Cell Models. Front. Pediatr. 2020, 8, 536. [Google Scholar] [CrossRef]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef]
- Calucho, M.; Gartner, S.; Barranco, P.; Fernández-Álvarez, P.; Pérez, R.G.; Tizzano, E.F. Validation of nasospheroids to assay CFTR functionality and modulator responses in cystic fibrosis. Sci. Rep. 2021, 11, 15511. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Q.; Sun, X.; Shen, J.; Chen, H. Organoids as a Powerful Model for Respiratory Diseases. Stem Cells Int. 2020, 2020, 5847876. [Google Scholar] [CrossRef]
- Cidem, A.; Bradbury, P.; Traini, D.; Ong, H.X. Modifying and Integrating in vitro and ex vivo Respiratory Models for Inhalation Drug Screening. Front. Bioeng. Biotechnol. 2020, 8, 581995. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.L.; Chan, Y.; Candasamy, M.; Chellian, J.; Madheswaran, T.; Sakthivel, L.P.; Patel, V.K.; Chakraborty, A.; MacLoughlin, R.; Kumar, D.; et al. Unravelling the molecular mechanisms underlying chronic respiratory diseases for the development of novel therapeutics via in vitro experimental models. Eur. J. Pharmacol. 2022, 919, 174821. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Anderson, J.D.; Deng, L.; Mackay, S.; Bailey, J.; Kersh, L.; Rowe, S.M.; Guimbellot, J.S. Human Nasal Epithelial Organoids for Therapeutic Development in Cystic Fibrosis. Genes 2020, 11, 603. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.; Bleich, M.; Schürlein, M.; Kühr, J.; Seydewitz, H.H.; Brandis, M.; Greger, R.; Kunzelmann, K. Cholinergic ion secretion in human colon requires coactivation by cAMP. Am. J. Physiol. Content 1998, 275, G1274–G1281. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Mall, M. Electrolyte Transport in the Mammalian Colon: Mechanisms and Implications for Disease. Physiol. Rev. 2002, 82, 245–289. [Google Scholar] [CrossRef]
- Mall, M.; Wissner, A.; Seydewitz, H.H.; Kuehr, J.; Brandis, M.; Greger, R.; Kunzelmann, K. Defective cholinergic Cl− secretion and detection of K+ secretion in rectal biopsies from cystic fibrosis patients. Am. J. Physiol. Liver Physiol. 2000, 278, G617–G624. [Google Scholar] [CrossRef] [PubMed]
- Veeze, H.J.; Sinaasappel, M.; Bijman, J.; Bouquet, J.; De Jonge, H.R. Ion transport abnormalities in rectal suction biopsies from children with cystic fibrosis. Gastroenterology 1991, 101, 398–403. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Vitzthum, C.; Mall, M.A. Potential of Intestinal Current Measurement for Personalized Treatment of Patients with Cystic Fibrosis. J. Pers. Med. 2021, 11, 384. [Google Scholar] [CrossRef]
- Veeze, H.J.; Halley, D.J.J.; Bijman, J.; de Jongste, J.C.; de Jonge, H.R.; Sinaasappel, M. Determinants of mild clinical symptoms in cystic fibrosis patients. Residual chloride secretion measured in rectal biopsies in relation to the genotype. J. Clin. Investig. 1994, 93, 461–466. [Google Scholar] [CrossRef]
- Derichs, N.; Sanz, J.; Von Kanel, T.; Stolpe, C.; Zapf, A.; Tümmler, B.; Gallati, S.; Ballmann, M. Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: Validation and reference data. Thorax 2010, 65, 594–599. [Google Scholar] [CrossRef]
- de Jonge, H.R.; Ballmann, M.; Veeze, H.; Bronsveld, I.; Stanke, F.; Tümmler, B.; Sinaasappel, M. Ex vivo CF diagnosis by intestinal current measurements (ICM) in small aperture, circulating Ussing chambers. J. Cyst. Fibros. 2004, 3, 159–163. [Google Scholar] [CrossRef]
- Mall, M.; Hirtz, S.; Gonska, T.; Kunzelmann, K. Assessment of CFTR function in rectal biopsies for the diagnosis of cystic fibrosis. J. Cyst. Fibros. 2004, 3, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.A.L.; Duarte, A.; Marson, F.A.L.; Centeio, R.; Doušová, T.; Kunzelmann, K.; Amaral, M.D. Assessment of Distinct Electrophysiological Parameters in Rectal Biopsies for the Choice of the Best Diagnosis/Prognosis Biomarkers for Cystic Fibrosis. Front. Physiol. 2020, 11, 604580. [Google Scholar] [CrossRef] [PubMed]
- Hirtz, S.; Gonska, T.; Seydewitz, H.H.; Thomas, J.; Greiner, P.; Kuehr, J.; Brandis, M.; Eichler, I.; Rocha, H.; Lopes, A.I.; et al. CFTR Cl− channel function in native human colon correlates with the genotype and phenotype in cystic fibrosis. Gastroenterology 2004, 127, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15.e1. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Hug, M.J.; Sommerburg, O.; Hirtz, S.; Hentschel, J.; Heinzmann, A.; Dopfer, C.; Schulz, A.; Mainz, J.G.; Tümmler, B.; et al. Intestinal Current Measurements Detect Activation of Mutant CFTR in Patients with Cystic Fibrosis with the G551D Mutation Treated with Ivacaftor. Am. J. Respir. Crit. Care Med. 2015, 192, 1252–1255. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Dopfer, C.; Naehrlich, L.; Gyulumyan, L.; Scheuermann, H.; Hirtz, S.; Wege, S.; Mairbäurl, H.; Dorda, M.; Hyde, R.; et al. Effects of Lumacaftor–Ivacaftor Therapy on Cystic Fibrosis Transmembrane Conductance Regulator Function in Phe508del Homozygous Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 1433–1442. [Google Scholar] [CrossRef]
- Masson, A.; Schneider-Futschik, E.K.; Baatallah, N.; Nguyen-Khoa, T.; Girodon, E.; Hatton, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; et al. Predictive factors for lumacaftor/ivacaftor clinical response. J. Cyst. Fibros. 2019, 18, 368–374. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Vitzhum, C.; Pallenber, S.T.; Naehrlich, L.; Stahl, M.; Rohrbach, A.; Drescher, M.; Minso, R.; Ringshausen, F.C.; Rueckes-Nilges, C.; et al. Effects of Elexacaftor/Tezacaftor/Ivacaftor Therapy on CFTR Function in Patients with Cystic Fibrosis and One or Two F508del Alleles. Am. J. Respir. Crit. Care Med. 2022, 205, 540–549. [Google Scholar] [CrossRef]
- Wilschanski, M.; Yaakov, Y.; Omari, I.; Zaman, M.; Martin, C.R.; Cohen-Cymberknoh, M.; Shoseyov, D.; Kerem, E.; Dasilva, D.; Sheth, S.; et al. Comparison of Nasal Potential Difference and Intestinal Current Measurements as Surrogate Markers for CFTR Function. J. Pediatr. Gastroenterol. Nutr. 2016, 63, e92–e97. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; De Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; De Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Vonk, A.M.; van Mourik, P.; Ramalho, A.S.; Silva, I.A.L.; Statia, M.; Kruisselbrink, E.; Suen, S.W.F.; Dekkers, J.F.; Vleggaar, F.P.; Houwen, R.H.J.; et al. Protocol for Application, Standardization and Validation of the Forskolin-Induced Swelling Assay in Cystic Fibrosis Human Colon Organoids. STAR Protoc. 2020, 1, 100019. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Gondra, R.A.G.; Kruisselbrink, E.; Vonk, A.M.; Janssens, H.M.; Groot, K.M.D.W.; Van Der Ent, C.K.; Beekman, J.M. Optimal correction of distinct CFTR folding mutants in rectal cystic fibrosis organoids. Eur. Respir. J. 2016, 48, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Maule, G.; Casini, A.; Montagna, C.; Ramalho, A.S.; De Boeck, K.; Debyser, Z.; Carlon, M.S.; Petris, G.; Cereseto, A. Allele specific repair of splicing mutations in cystic fibrosis through AsCas12a genome editing. Nat. Commun. 2019, 10, 3556. [Google Scholar] [CrossRef]
- Geurts, M.H.; de Poel, E.; Amatngalim, G.D.; Oka, R.; Meijers, F.M.; Kruisselbrink, E.; van Mourik, P.; Berkers, G.; de Winter-de Groot, K.M.; Michel, S.; et al. CRISPRBased Adenine Editors Correct Nonsense Mutations in a Cystic Fibrosis Organoid Biobank. Cell Stem Cell 2020, 26, 503–510.e7. [Google Scholar] [CrossRef]
- Silva, I.A.L.; Railean, V.; Duarte, A.; Amaral, M.D. Personalized Medicine Based on Nasal Epithelial Cells: Comparative Studies with Rectal Biopsies and Intestinal Organoids. J. Pers. Med. 2021, 11, 421. [Google Scholar] [CrossRef] [PubMed]
- Collaco, J.M.; Blackman, S.M.; Raraigh, K.S.; Corvol, H.; Rommens, J.M.; Pace, R.G.; Boelle, P.Y.; McGready, J.; Sosnay, P.R.; Strug, L.J.; et al. Sources of Variation in Sweat Chloride Measurements in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 194, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- de Winter-de Groot, K.M.; Janssens, H.M.; Van Uum, R.T.; Dekkers, J.F.; Berkers, G.; Vonk, A.; Kruisselbrink, E.; Oppelaar, H.; Vries, R.; Clevers, H.; et al. Stratifying infants with cystic fibrosis for disease severity using intestinal organoid swelling as a biomarker of CFTR function. Eur. Respir. J. 2018, 52, 1702529. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Accurso, F.; Clancy, J.P. Detection of Cystic Fibrosis Transmembrane Conductance Regulator Activity in Early-Phase Clinical Trials. Proc. Am. Thorac. Soc. 2007, 4, 387–398. [Google Scholar] [CrossRef]
- Kyrilli, S.; Henry, T.; Wilschanski, M.; Fajac, I.; Davies, J.C.; Jais, J.P.; Sermet-Gaudelus, I. Insights into the variability of nasal potential difference, a biomarker of CFTR activity. J. Cyst. Fibros. 2020, 19, 620–626. [Google Scholar] [CrossRef]
- Muilwijk, D.; de Poel, E.; van Mourik, P.; Suen, S.W.F.; Vonk, A.M.; Brunsveld, J.E.; Kruisselbrink, E.; Oppelaar, H.; Hagemeijer, M.C.; Berkers, G.; et al. Forskolin-induced Organoid Swelling is Associated with Long-term CF Disease Progression. Eur. Respir. J. 2022, 2100508. [Google Scholar] [CrossRef]
- de Poel, E.; Spelier, S.; Suen, S.W.F.; Kruisselbrink, E.; Graeber, S.Y.; Mall, M.A.; Weersink, E.J.M.; van der Eerden, M.M.; Koppelman, G.H.; van der Ent, C.K.; et al. Functional Restoration of CFTR Nonsense Mutations in Intestinal Organoids. J. Cyst. Fibros. 2022, 21, 246–253. [Google Scholar] [CrossRef] [PubMed]
- de Winter–de Groot, K.M.; Berkers, G.; Marck–van der Wilt, R.E.P.; van der Meer, R.; Vonk, A.; Dekkers, J.F.; Geerdink, M.; Michel, S.; Kruisselbrink, E.; Vries, R.; et al. Forskolin-induced swelling of intestinal organoids correlates with disease severity in adults with cystic fibrosis and homozygous F508del mutations. J. Cyst. Fibros. 2020, 19, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, A.S.; Furstova, E.; Vonk, A.M.; Ferrante, M.; Verfailli, C.; Dupont, L.; Boon, M.; Proesmans, M.; Beekma, J.M.; Sarouk, I.; et al. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur. Respir. J. 2021, 57, 1902426. [Google Scholar] [CrossRef] [PubMed]
- Aalbers, B.L.; Brunsveld, J.E.; van der Ent, C.K.; van den Eijnden, J.C.; Beekman, J.M.; Heijerman, H.G.M. Forskolin induced swelling (FIS) assay in intestinal organoids to guide eligibility for compassionate use treatment in a CF patient with a rare genotype. J. Cyst. Fibros. 2022, 21, 254–257. [Google Scholar] [CrossRef]
- Graeber, S.Y.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Hirtz, S.; van der Ent, C.K.; Mall, M.A.; Beekman, J.M. Comparison of Organoid Swelling and In Vivo Biomarkers of CFTR Function to Determine Effects of Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation. Am. J. Respir. Crit. Care Med. 2020, 202, 1589–1592. [Google Scholar] [CrossRef]
- Zomer-van Ommen, D.D.; de Poel, E.; Kruisselbrink, E.; Oppelaar, H.; Vonk, A.M.; Janssens, H.M.; van der Ent, C.K.; Hagemeijer, M.C.; Beekman, J.M. Comparison of ex vivo and in vitro intestinal cystic fibrosis models to measure CFTR-dependent ion channel activity. J. Cyst. Fibros. 2018, 17, 316–324. [Google Scholar] [CrossRef]
- Ciciriello, F.; Bijvelds, M.J.C.; Alghisi, F.; Meijsen, K.F.; Cristiani, L.; Sorio, C.; Melotti, P.; Fiocchi, A.G.; Lucidi, V.; De Jonge, H.R. Theratyping of the Rare CFTR Variants E193K and R334W in Rectal Organoid-Derived Epithelial Monolayers. J. Pers. Med. 2022, 12, 632. [Google Scholar] [CrossRef]
- Ferrera, L.; Baroni, D.; Moran, O. Lumacaftor-rescued F508del-CFTR has a modified bicarbonate permeability. J. Cyst. Fibros. 2019, 18, 602–605. [Google Scholar] [CrossRef]
- Fiore, M.; Picco, C.; Moran, O. Correctors modify the bicarbonate permeability of F508del-CFTR. Sci. Rep. 2020, 10, 8440. [Google Scholar] [CrossRef]
- Foulke-Abel, J.; In, J.; Yin, J.; Zachos, N.C.; Kovbasnjuk, O.; Estes, M.K.; de Jonge, H.; Donowitz, M. Human Enteroids as a Model of Upper Small Intestinal Ion Transport Physiology and Pathophysiology. Gastroenterology 2016, 150, 638–649.e8. [Google Scholar] [CrossRef] [PubMed]
- Al Abbar, A.; Ngai, S.C.; Nograles, N.; Alhaji, S.Y.; Abdullah, S. Induced Pluripotent Stem Cells: Reprogramming Platforms and Applications in Cell Replacement Therapy. BioResearch Open Access 2020, 9, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Ensinck, M.; Mottais, A.; Detry, C.; Leal, T.; Carlon, M.S. On the Corner of Models and Cure: Gene Editing in Cystic Fibrosis. Front. Pharmacol. 2021, 12, 662110. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.P.; Bear, C.E.; Chin, S.; Pasceri, P.; Thompson, T.O.; Huan, L.J.; Ratjen, F.; Ellis, J.; Rossant, J. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat. Biotechnol. 2012, 30, 876–882. [Google Scholar] [CrossRef] [PubMed]
- McCauley, K.B.; Hawkins, F.; Serra, M.; Thomas, D.C.; Jacob, A.; Kotton, D.N. Efficient Derivation of Functional Human Airway Epithelium from Pluripotent Stem Cells via Temporal Regulation of Wnt Signaling. Cell Stem Cell 2017, 20, 844–857.e6. [Google Scholar] [CrossRef]
- Huang, S.X.L.; Islam, M.N.; O’Neill, J.; Hu, Z.; Yang, Y.G.; Chen, Y.W.; Mumau, M.; Green, M.D.; Vunjak-Novakovic, G.; Bhattacharya, J.; et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 84–91. [Google Scholar] [CrossRef]
- Firth, A.L.; Dargitz, C.T.; Qualls, S.J.; Menon, T.; Wright, R.; Singer, O.; Gage, F.H.; Khanna, A.; Verma, I.M. Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2014, 111, E1723–E1730. [Google Scholar] [CrossRef]
- Hawkins, F.; Kramer, P.; Jacob, A.; Driver, I.; Thomas, D.C.; McCauley, K.B.; Skvir, N.; Crane, A.M.; Kurmann, A.A.; Hollenberg, A.N.; et al. Prospective isolation of NKX2-1–expressing human lung progenitors derived from pluripotent stem cells. J. Clin. Investig. 2017, 127, 2277–2294. [Google Scholar] [CrossRef]
- Merkert, S.; Schubert, M.; Olmer, R.; Engels, L.; Radetzki, S.; Veltman, M.; Scholte, B.J.; Zöllner, J.; Pedemonte, N.; Galietta, L.J.V.; et al. High-Throughput Screening for Modulators of CFTR Activity Based on Genetically Engineered Cystic Fibrosis Disease-Specific iPSCs. Stem Cell Rep. 2019, 12, 1389–1403. [Google Scholar] [CrossRef]
- Ahmadi, S.; Bozoky, Z.; Di Paola, M.; Xia, S.; Li, C.; Wong, A.P.; Wellhauser, L.; Molinski, S.V.; Ip, W.; Ouyang, H.; et al. Phenotypic profiling of CFTR modulators in patient-derived respiratory epithelia. NPJ Genom. Med. 2017, 2, 12. [Google Scholar] [CrossRef]
- Crane, A.M.; Kramer, P.; Bui, J.H.; Chung, W.J.; Li, X.S.; Gonzalez-Garay, M.L.; Hawkins, F.; Liao, W.; Mora, D.; Choi, S.; et al. Targeted Correction and Restored Function of the CFTR Gene in Cystic Fibrosis Induced Pluripotent Stem Cells. Stem Cell Rep. 2015, 4, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Wellhauser, L.; Laselva, O.; Utkina, I.; Bozoky, Z.; Gunawardena, T.; Ngan, Z.; Xia, S.; Di Paola, M.; Eckford, P.D.W.; et al. A new platform for high-throughput therapy testing on iPSC-derived lung progenitor cells from cystic fibrosis patients. Stem Cell Rep. 2021, 16, 2825–2837. [Google Scholar] [CrossRef]
- Berical, A.; Lee, R.E.; Lu, J.; Beermann, M.L.; Le Suer, J.A.; Mithal, A.; Thomas, D.; Ranallo, N.; Peasley, M.; Stuffer, A.; et al. A multimodal iPSC platform for cystic fibrosis drug testing. Nat. Commun. 2022, 13, 4270. [Google Scholar] [CrossRef]
- Ngan, S.Y.; Quach, H.T.; Laselva, O.; Huang, E.N.; Mangos, M.; Xia, S.; Bear, C.E.; Wong, A.P. Stage-Specific Generation of Human Pluripotent Stem Cell Derived Lung Models to Measure CFTR Function. Curr. Protoc. 2022, 2, e341. [Google Scholar] [CrossRef] [PubMed]
- Erwood, S.; Laselva, O.; Bily, T.M.; Brewer, R.A.; Rutherford, A.H.; Bear, C.E.; Ivakine, E.A. Allele-Specific Prevention of Nonsense-Mediated Decay in Cystic Fibrosis Using Homology-Independent Genome Editing. Mol. Ther. Methods Clin. Dev. 2020, 17, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Smirnikhina, S.A.; Kondrateva, E.V.; Adilgereeva, E.P.; Anuchina, A.A.; Zaynitdinova, M.I.; Slesarenko, Y.S.; Ershova, A.S.; Ustinov, K.D.; Yasinovsky, M.I.; Amelina, E.L.; et al. P.F508del editing in cells from cystic fibrosis patients. PLoS ONE 2020, 15, e0242094. [Google Scholar] [CrossRef]
- Ruan, J.; Hirai, H.; Yang, D.; Ma, L.; Hou, X.; Jiang, H.; Wei, H.; Rajagopalan, C.; Mou, H.; Wang, G.; et al. Efficient Gene Editing at Major CFTR Mutation Loci. Mol. Ther. Nucleic Acids 2019, 16, 73–81. [Google Scholar] [CrossRef]
- Firth, A.L.; Menon, T.; Parker, G.S.; Qualls, S.J.; Lewis, B.M.; Ke, E.; Dargitz, C.T.; Wright, R.; Khanna, A.; Gage, F.H.; et al. Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. Cell Rep. 2015, 12, 1385–1390. [Google Scholar] [CrossRef]
- Suzuki, S.; Sargent, R.G.; Illek, B.; Fischer, H.; Esmaeili-Shandiz, A.; Yezzi, M.J.; Lee, A.; Yang, Y.; Kim, S.; Renz, P.; et al. TALENs Facilitate Single-step Seamless SDF Correction of F508del CFTR in Airway Epithelial Submucosal Gland Cell-derived CF-iPSCs. Mol. Ther. Nucleic Acids 2016, 5, e273. [Google Scholar] [CrossRef]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–109. [Google Scholar] [CrossRef]
- Merkert, S.; Bednarski, C.; Göhring, G.; Cathomen, T.; Martin, U. Generation of a gene-corrected isogenic control iPSC line from cystic fibrosis patient-specific iPSCs homozygous for p.Phe508del mutation mediated by TALENs and ssODN. Stem Cell Res. 2017, 23, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Mithal, A.; Capilla, A.; Heinze, D.; Berical, A.; Villacorta-Martin, C.; Vedaie, M.; Jacob, A.; Abo, K.; Szymaniak, A.; Peasley, M.; et al. Generation of mesenchyme free intestinal organoids from human induced pluripotent stem cells. Nat. Commun. 2020, 11, 215. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.L.; Mahe, M.M.; Múnera, J.; Howell, J.C.; Sundaram, N.; Poling, H.M.; Schweitzer, J.I.; Vallance, J.E.; Mayhew, C.N.; Sun, Y.; et al. An in vivo model of human small intestine using pluripotent stem cells. Nat. Med. 2014, 20, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Bozóky, Z.; Di Paola, M.; Laselva, O.; Ahmadi, S.; Jiang, J.X.; Pitstick, A.L.; Jiang, C.; Rotin, D.; Mayhew, C.N.; et al. High-Throughput Functional Analysis of CFTR and Other Apically Localized Proteins in iPSCDerived Human Intestinal Organoids. Cells 2021, 10, 3419. [Google Scholar] [CrossRef]
- Hohwieler, M.; Illing, A.; Hermann, P.C.; Mayer, T.; Stockmann, M.; Perkhofer, L.; Eiseler, T.; Antony, J.S.; Müller, M.; Renz, S.; et al. Human pluripotent stem cell-derived acinar/ductal organoids generate human pancreas upon orthotopic transplantation and allow disease modelling. Gut 2017, 66, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Ogawa, S.; E Bear, C.; Ahmadi, S.; Chin, S.; Li, B.; Grompe, M.; Keller, G.; Kamath, B.M.; Ghanekar, A. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat. Biotechnol. 2015, 33, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Dianat, N.; Dubois-Pot-Schneider, H.; Steichen, C.; Desterke, C.; Leclerc, P.; Raveux, A.; Combettes, L.; Weber, A.; Corlu, A.; Dubart-Kupperschmitt, A. Generation of functional cholangiocyte-like cells from human pluripotent stem cells and HepaRG cells. Hepatology 2014, 60, 700–714. [Google Scholar] [CrossRef]
- Fiorotto, R.; Amenduni, M.; Mariotti, V.; Fabris, L.; Spirli, C.; Strazzabosco, M. Src kinase inhibition reduces inflammatory and cytoskeletal changes in ΔF508 human cholangiocytes and improves cystic fibrosis transmembrane conductance regulator correctors efficacy. Hepatology 2018, 67, 972–988. [Google Scholar] [CrossRef]
- Ogawa, M.; Jiang, J.X.; Xia, S.; Yang, D.; Ding, A.; Laselva, O.; Hernandez, M.; Cui, C.; Higuchi, Y.; Suemizu, H.; et al. Generation of functional ciliated cholangiocytes from human pluripotent stem cells. Nat. Commun. 2021, 12, 6504. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Yuan Hsin, H.; Ingber, D.E. Reconstituting Organ-Level Lung Functions on a Chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-chip: Recent breakthroughs and future prospects. Biomed. Eng. Online 2020, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Konar, D.; Devarasetty, M.; Yildiz, D.V.; Atala, A.; Murphy, S.V. Lung-On-AChip Technologies for Disease Modeling and Drug Development. Biomed. Eng. Comput. Biol. 2016, 7, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Mejías, J.C.; Nelson, M.R.; Liseth, O.; Roy, K. A 96-well format microvascularized human lung-on-a-chip platform for microphysiological modeling of fibrotic diseases. Lab Chip 2020, 20, 3601–3611. [Google Scholar] [CrossRef] [PubMed]
- Plebani, R.; Potla, R.; Soong, M.; Bai, H.; Izadifar, Z.; Jiang, A.; Travis, R.N.; Belgur, C.; Dinis, A.; Cartwright, M.J.; et al. Modeling pulmonary cystic fibrosis in a human lung airway-on-a-chip: Cystic fibrosis airway chip. J. Cyst. Fibros. 2021, 21, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Benam, K.H.; Villenave, R.; Lucchesi, C.; Varone, A.; Hubeau, C.; Lee, H.H.; E Alves, S.; Salmon, M.; Ferrante, T.C.; Weaver, J.C.; et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat. Methods 2015, 13, 151–157. [Google Scholar] [CrossRef]
- Nawroth, J.C.; Lucchesi, C.; Cheng, D.; Shukla, A.; Ngyuen, J.; Shroff, T.; Varone, A.; Karalis, K.; Lee, H.H.; Alves, S.; et al. A Microengineered Airway Lung Chip Models Key Features of Viral-induced Exacerbation of Asthma. Am. J. Respir. Cell Mol. Biol. 2020, 63, 591–600. [Google Scholar] [CrossRef]
- Lavelle, G.M.; White, M.M.; Browne, N.; McElvaney, N.G.; Reeves, E.P. Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences. BioMed Res. Int. 2016, 2016, 5258727. [Google Scholar] [CrossRef]
- Semaniakou, A.; Croll, R.P.; Chappe, V. Animal Models in the Pathophysiology of Cystic Fibrosis. Front. Pharmacol. 2019, 9, 1475. [Google Scholar] [CrossRef]
- Shik Mun, K.; Arora, K.; Huang, Y.; Yang, F.; Yarlagadda, S.; Ramananda, Y.; Abu-El-Haija, M.; Palermo, J.J.; Appakalai, B.N.; Nathan, J.D.; et al. Patient-derived pancreas-on-a-chip to model cystic fibrosis-related disorders. Nat. Commun. 2019, 10, 3124. [Google Scholar] [CrossRef]
- Fawcett, L.K.; Wakefield, C.E.; Sivam, S.; Middleton, P.G.; Wark, P.; Widger, J.; Jaffe, A.; Waters, S.A. Avatar acceptability: Views from the Australian Cystic Fibrosis community on the use of personalised organoid technology to guide treatment decisions. ERJ Open Res. 2021, 7, 00448–02020. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cell Type | Examples | Most Frequent Uses |
|---|---|---|
| Non-human non-epithelial | BHK, 3T3 | Extensively used in initial HTS assays Characterization of CFTR variants and CFTR biology |
| CHO, Cos-7 | Characterization of CFTR biology Assessment of common and rare CFTR variants (transient expression) | |
| Human non-epithelial | HEK | Assessment of common and rare CFTR variants (transient and stable expression) |
| Non-human epithelial | MDCK | Characterization of CFTR biology |
| FRT | Extensively used in HTS assays Assessment of common and rare CFTR variants (transient and stable expression) Label extension of clinically approved CFTR modulators (FDA only) | |
| Human epithelial | CFBE41o− | Extensively used in HTS assays Characterization of CFTR biology Assessment of common and rare CFTR variants (transient and stable expression) |
| 16HBE14o− | Assessment of common and rare CFTR variants in the native genomic context |
| Model | Advantages | Limitations |
|---|---|---|
| HBE cells in monolayer culture | Well-established protocols to assess efficacy of CFTR modulators Recapitulate lung disease in CF Assessment of downstream consequences of CFTR dysfunction (e.g., mucociliary transport rate) Potential usage to investigate modulation of alternative channels/transporters | Requirement of invasive procedures to be harvested Low availability/accessibility, particularly for rare CF genotypes Low throughput/limited scalability to assess multiple compounds (or combinations thereof) |
| HNE cells in monolayer culture | Minimal invasive procedures to be harvested HNE cells can be used as surrogates for HBE cells in CFTR studies Reasonable availability/accessibility, particularly for rare CF genotypes Potential usage to investigate modulation of alternative channels/transporters or downstream consequences of CFTR dysfunction Results from HNE cultures correlate well with in vivo biomarkers/clinical features | Low throughput/limited scalability to assess multiple compounds (or combinations thereof) Lack of standardized procedures |
| Airway organoids | Cultures achieve maturity and readiness for usage faster than ALI cultures Greater throughput/scalability than ALI cultures to assess multiple compounds | Requirement of invasive procedures to be harvested if derived from HBE cells Greater variability in results compared to ALI cultures Unfavorable response-to-background ratio Lack of standardized procedures |
| Rectal biopsies | Abundant expression of CFTR in distal colon tissue ICM is a sensitive biomarker of CFTR function | Requirement of invasive procedures (although with no to low pain associated) Requirement of testing all samples on the harvesting day (i.e., cannot be biobanked) Possible reduced penetration of CFTR modulators under ex vivo conditions |
| Intestinal organoids | High throughput/scalability to assess multiple compounds (or combinations thereof) Reasonable availability/accessibility, particularly for rare CF genotypes Results from FIS assay correlate well with in vivo biomarkers/clinical features | Requirement of invasive procedures, since they are derived from rectal biopsies Unclear potential to investigate modulation of alternative channels/transporters |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, I.A.L.; Laselva, O.; Lopes-Pacheco, M. Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis. J. Pers. Med. 2022, 12, 1321. https://doi.org/10.3390/jpm12081321
Silva IAL, Laselva O, Lopes-Pacheco M. Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis. Journal of Personalized Medicine. 2022; 12(8):1321. https://doi.org/10.3390/jpm12081321
Chicago/Turabian StyleSilva, Iris A. L., Onofrio Laselva, and Miquéias Lopes-Pacheco. 2022. "Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis" Journal of Personalized Medicine 12, no. 8: 1321. https://doi.org/10.3390/jpm12081321