
Elexacaftor/Tezacaftor/Ivacaftor Accelerates Wound Repair in Cystic Fibrosis Airway Epithelium

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Wound Repair Assay
2.3. Elexacaftor/Tezacaftor/Ivacaftor Treatment
2.4. Evaluation of CFTR Function and Protein Maturation
2.4.1. CFTR Channel Function in CFBE-F508del
2.4.2. Immunoblotting
2.5. Statistical Analysis
3. Results
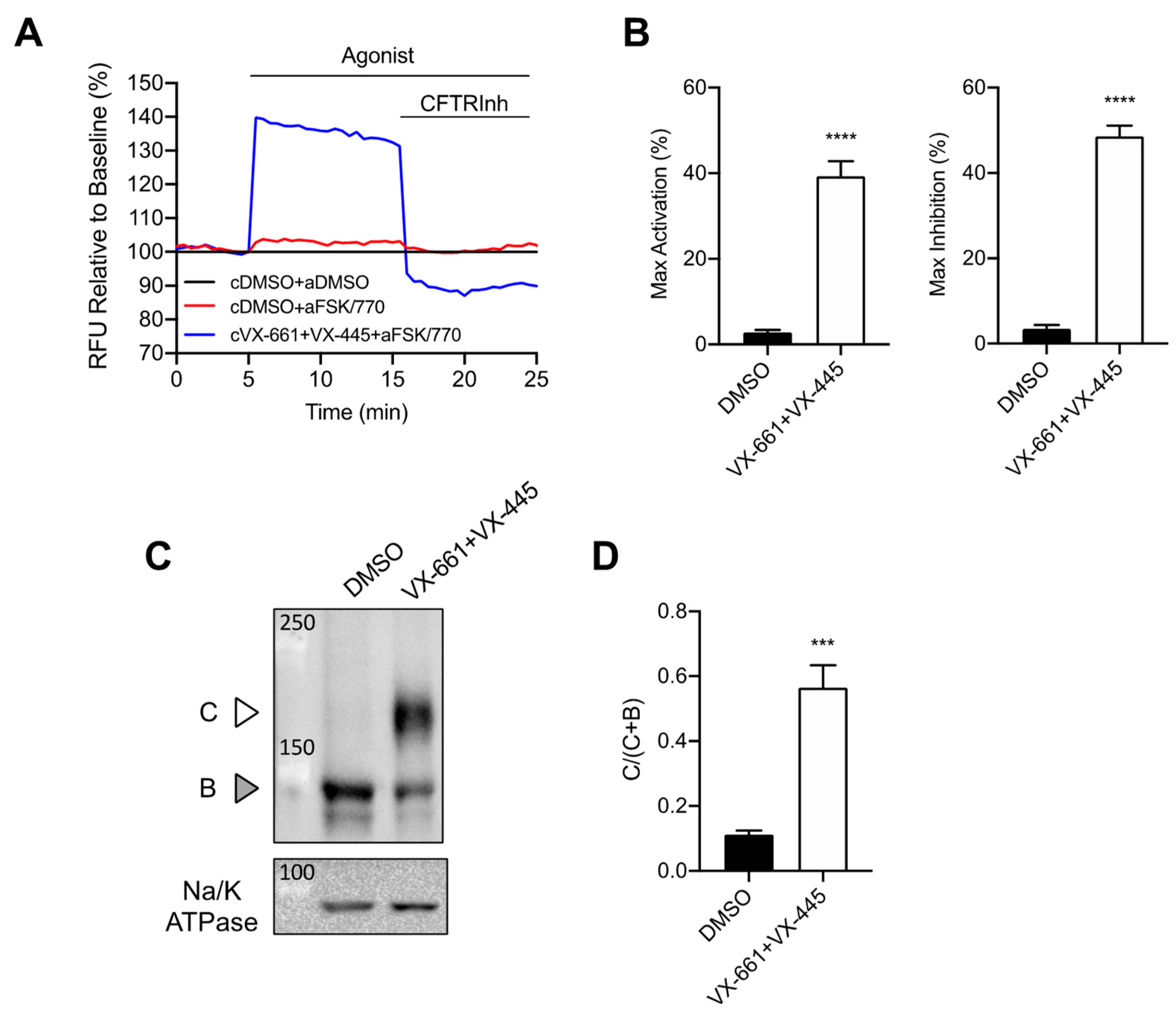
3.1. Rescue of CFTR F508del Maturation and Activity by Elexacaftor/Tezacaftor/Ivacaftor
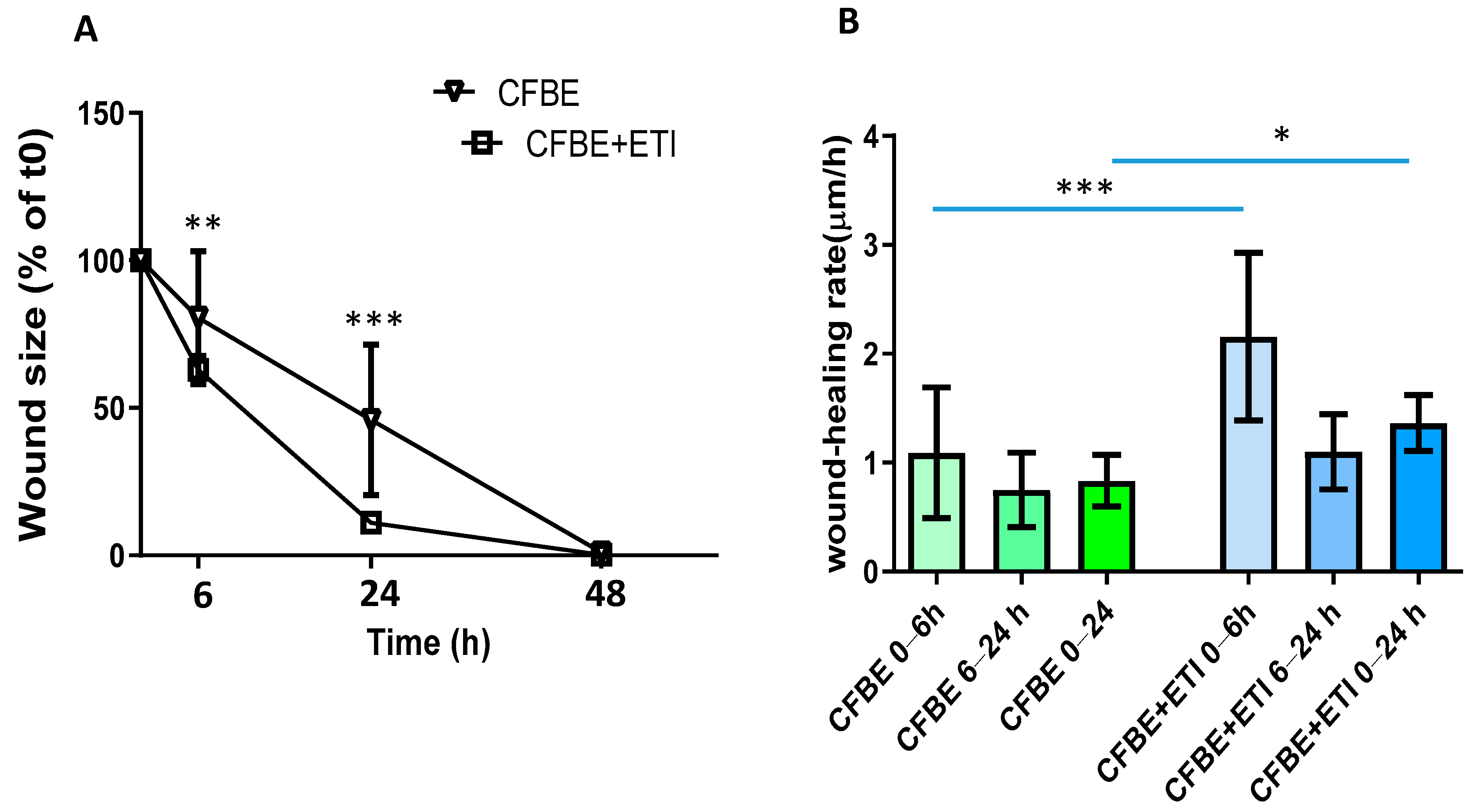
3.2. Effect of Elexacaftor/Tezacaftor/Ivacaftor on Wound Closure and Wound Healing Rate
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- CFTR2. Clinical and Functional Translation of CFTR. Available online: https://cftr2.org/ (accessed on 20 August 2022).
- Boyle, M.P.; De Boeck, K. A new era in the treatment of cystic fibrosis: Correction of the underlying CFTR defect. Lancet Respir. Med. 2013, 1, 158–163. [Google Scholar] [CrossRef]
- Farinha, C.M.; Matos, P. Repairing the basic defect in cystic fibrosis-one approach is not enough. FEBS J. 2016, 283, 246–264. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef]
- Connett, G.J. Lumacaftor-ivacaftor in the treatment of cystic fibrosis: Design, development and place in therapy. Drug Des. Devel. Ther. 2019, 13, 2405–2412. [Google Scholar] [CrossRef]
- Aalbers, B.L.; de Winter-de Groot, K.M.; Arets, H.G.M.; Hofland, R.W.; de Kiviet, A.C.; van Oirschot-van de Ven, M.M.M.; Kruijswijk, M.A.; Schotman, S.; Michel, S.; van der Ent, C.K.; et al. Clinical effect of lumacaftor/ivacaftor in F508del homozygous CF patients with FEV1 >/= 90% predicted at baseline. J. Cyst. Fibros. 2020, 19, 654–658. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Hubeau, C.; Lorenzato, M.; Couetil, J.P.; Hubert, D.; Dusser, D.; Puchelle, E.; Gaillard, D. Quantitative analysis of inflammatory cells infiltrating the cystic fibrosis airway mucosa. Clin. Exp. Immuno.l 2001, 124, 69–76. [Google Scholar] [CrossRef]
- Voynow, J.A.; Fischer, B.M.; Roberts, B.C.; Proia, A.D. Basal-like cells constitute the proliferating cell population in cystic fibrosis airways. Am. J. Respir. Crit. Care Med. 2005, 172, 1013–1018. [Google Scholar] [CrossRef] [Green Version]
- Leigh, M.W.; Kylander, J.E.; Yankaskas, J.R.; Boucher, R.C. Cell proliferation in bronchial epithelium and submucosal glands of cystic fibrosis patients. Am. J. Respir. Cell Mol. Biol. 1995, 12, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Adam, D.; Roux-Delrieu, J.; Luczka, E.; Bonnomet, A.; Lesage, J.; Merol, J.C.; Polette, M.; Abely, M.; Coraux, C. Cystic fibrosis airway epithelium remodelling: Involvement of inflammation. J. Pathol. 2015, 235, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Piorunek, T.; Marszalek, A.; Biczysko, W.; Gozdzik, J.; Cofta, S.; Seget, M. Correlation between the stage of cystic fibrosis and the level of morphological changes in adult patients. J. Physiol. Pharmacol. 2008, 59 (Suppl. 6), 565–572. [Google Scholar]
- Tiddens, H.A.; Koopman, L.P.; Lambert, R.K.; Elliott, W.M.; Hop, W.C.; van der Mark, T.W.; de Boer, W.J.; de Jongste, J.C. Cartilaginous airway wall dimensions and airway resistance in cystic fibrosis lungs. Eur. Respir. J. 2000, 15, 735–742. [Google Scholar] [CrossRef]
- Dovey, M.; Wisseman, C.L.; Roggli, V.L.; Roomans, G.M.; Shelburne, J.D.; Spock, A. Ultrastructural morphology of the lung in cystic fibrosis. J. Submicrosc. Cytol. Pathol. 1989, 21, 521–534. [Google Scholar]
- Durieu, I.; Peyrol, S.; Gindre, D.; Bellon, G.; Durand, D.V.; Pacheco, Y. Subepithelial fibrosis and degradation of the bronchial extracellular matrix in cystic fibrosis. Am. J. Respir. Crit. Care Med. 1998, 158, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D.; Quaresma, M.C.; Pankonien, I. What Role Does CFTR Play in Development, Differentiation, Regeneration and Cancer? Int. J. Mol. Sci. 2020, 21, 3133. [Google Scholar] [CrossRef]
- Adam, D.; Bilodeau, C.; Sognigbe, L.; Maille, E.; Ruffin, M.; Brochiero, E. CFTR rescue with VX-809 and VX-770 favors the repair of primary airway epithelial cell cultures from patients with class II mutations in the presence of Pseudomonas aeruginosa exoproducts. J. Cyst. Fibros. 2018, 17, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Criscione, M.L.; Allegretta, C.; Di Gioia, S.; Liso, A.; Conese, M. Insulin-Like Growth Factor Binding Protein (IGFBP-6) as a Novel Regulator of Inflammatory Response in Cystic Fibrosis Airway Cells. Front. Mol. Biosci. 2022, 9, 905468. [Google Scholar] [CrossRef]
- Trinh, N.T.; Bardou, O.; Prive, A.; Maille, E.; Adam, D.; Lingee, S.; Ferraro, P.; Desrosiers, M.Y.; Coraux, C.; Brochiero, E. Improvement of defective cystic fibrosis airway epithelial wound repair after CFTR rescue. Eur. Respir. J. 2012, 40, 1390–1400. [Google Scholar] [CrossRef]
- Beccia, E.; Daniello, V.; Laselva, O.; Leccese, G.; Mangiacotti, M.; Di Gioia, S.; La Bella, G.; Guerra, L.; Matteo, M.; Angiolillo, A.; et al. Human Amniotic Mesenchymal Stem Cells and Fibroblasts Accelerate Wound Repair of Cystic Fibrosis Epithelium. Life 2022, 12, 756. [Google Scholar] [CrossRef] [PubMed]
- Erwood, S.; Laselva, O.; Bily, T.M.I.; Brewer, R.A.; Rutherford, A.H.; Bear, C.E.; Ivakine, E.A. Allele-Specific Prevention of Nonsense-Mediated Decay in Cystic Fibrosis Using Homology-Independent Genome Editing. Mol. Ther. Methods Clin. Dev. 2020, 17, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Bartlett, C.; Popa, A.; Ouyang, H.; Gunawardena, T.N.A.; Gonska, T.; Moraes, T.J.; Bear, C.E. Emerging preclinical modulators developed for F508del-CFTR have the potential to be effective for ORKAMBI resistant processing mutants. J. Cyst. Fibros. 2021, 20, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Chin, S.; Ramjeesingh, M.; Hung, M.; Ereno-Oreba, J.; Cui, H.; Laselva, O.; Julien, J.P.; Bear, C.E. Cholesterol Interaction Directly Enhances Intrinsic Activity of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). Cells 2019, 8, 804. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of multiple class II CFTR mutations by elexacaftor+ tezacaftor+ivacaftor mediated in part by the dual activities of Elexacaftor as both corrector and potentiator. Eur. Respir. J. 2021, 57, 2002774. [Google Scholar] [CrossRef]
- Laselva, O.; Guerra, L.; Castellani, S.; Favia, M.; Di Gioia, S.; Conese, M. Small-molecule drugs for cystic fibrosis: Where are we now? Pulm. Pharmacol. Ther. 2022, 72, 102098. [Google Scholar] [CrossRef]
- Mergiotti, M.; Murabito, A.; Prono, G.; Ghigo, A. CFTR Modulator Therapy for Rare CFTR Mutants. J. Respir. 2022, 2, 59–76. [Google Scholar] [CrossRef]
- Conese, M.; Di Gioia, S. Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis. Pathophysiology 2021, 28, 155–188. [Google Scholar] [CrossRef]
- Trinh, N.T.; Prive, A.; Maille, E.; Noel, J.; Brochiero, E. EGF and K+ channel activity control normal and cystic fibrosis bronchial epithelia repair. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L866–L880. [Google Scholar] [CrossRef]
- Schiller, K.R.; Maniak, P.J.; O’Grady, S.M. Cystic fibrosis transmembrane conductance regulator is involved in airway epithelial wound repair. Am. J. Physiol. Cell Physiol. 2010, 299, C912–C921. [Google Scholar] [CrossRef] [PubMed]
- Maille, E.; Trinh, N.T.; Prive, A.; Bilodeau, C.; Bissonnette, E.; Grandvaux, N.; Brochiero, E. Regulation of normal and cystic fibrosis airway epithelial repair processes by TNF-alpha after injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L945–L955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itokazu, Y.; Pagano, R.E.; Schroeder, A.S.; O’Grady, S.M.; Limper, A.H.; Marks, D.L. Reduced GM1 ganglioside in CFTR-deficient human airway cells results in decreased beta1-integrin signaling and delayed wound repair. Am. J. Physiol. Cell Physiol. 2014, 306, C819–C830. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, M.C.; Pankonien, I.; Clarke, L.A.; Sousa, L.S.; Silva, I.A.L.; Railean, V.; Dousova, T.; Fuxe, J.; Amaral, M.D. Mutant CFTR Drives TWIST1 mediated epithelial-mesenchymal transition. Cell Death Dis. 2020, 11, 920. [Google Scholar] [CrossRef]
- Sousa, L.; Pankonien, I.; Simoes, F.B.; Chanson, M.; Amaral, M.D. Impact of KLF4 on Cell Proliferation and Epithelial Differentiation in the Context of Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 6717. [Google Scholar] [CrossRef]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef]
- Hudson, R.P.; Dawson, J.E.; Chong, P.A.; Yang, Z.; Millen, L.; Thomas, P.J.; Brouillette, C.G.; Forman-Kay, J.D. Direct Binding of the Corrector VX-809 to Human CFTR NBD1: Evidence of an Allosteric Coupling between the Binding Site and the NBD1:CL4 Interface. Mol. Pharmacol. 2017, 92, 124–135. [Google Scholar] [CrossRef]
- Laselva, O.; Qureshi, Z.; Zeng, Z.W.; Petrotchenko, E.V.; Ramjeesingh, M.; Hamilton, C.M.; Huan, L.J.; Borchers, C.H.; Pomes, R.; Young, R.; et al. Identification of binding sites for ivacaftor on the cystic fibrosis transmembrane conductance regulator. iScience 2021, 24, 102542. [Google Scholar] [CrossRef]
- Laselva, O.; Molinski, S.; Casavola, V.; Bear, C.E. Correctors of the Major Cystic Fibrosis Mutant Interact through Membrane-Spanning Domains. Mol. Pharmacol. 2018, 93, 612–618. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR correction by type I folding correctors. Cell 2022, 185, 158–168.e111. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, A.; Prono, G.; Riccardi, E.; De Rose, V. Dysfunctional Inflammation in Cystic Fibrosis Airways: From Mechanisms to Novel Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 1952. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laselva, O.; Conese, M. Elexacaftor/Tezacaftor/Ivacaftor Accelerates Wound Repair in Cystic Fibrosis Airway Epithelium. J. Pers. Med. 2022, 12, 1577. https://doi.org/10.3390/jpm12101577
Laselva O, Conese M. Elexacaftor/Tezacaftor/Ivacaftor Accelerates Wound Repair in Cystic Fibrosis Airway Epithelium. Journal of Personalized Medicine. 2022; 12(10):1577. https://doi.org/10.3390/jpm12101577
Chicago/Turabian StyleLaselva, Onofrio, and Massimo Conese. 2022. "Elexacaftor/Tezacaftor/Ivacaftor Accelerates Wound Repair in Cystic Fibrosis Airway Epithelium" Journal of Personalized Medicine 12, no. 10: 1577. https://doi.org/10.3390/jpm12101577