
Non-Covalent BTK Inhibitors—The New BTKids on the Block for B-Cell Malignancies


Abstract
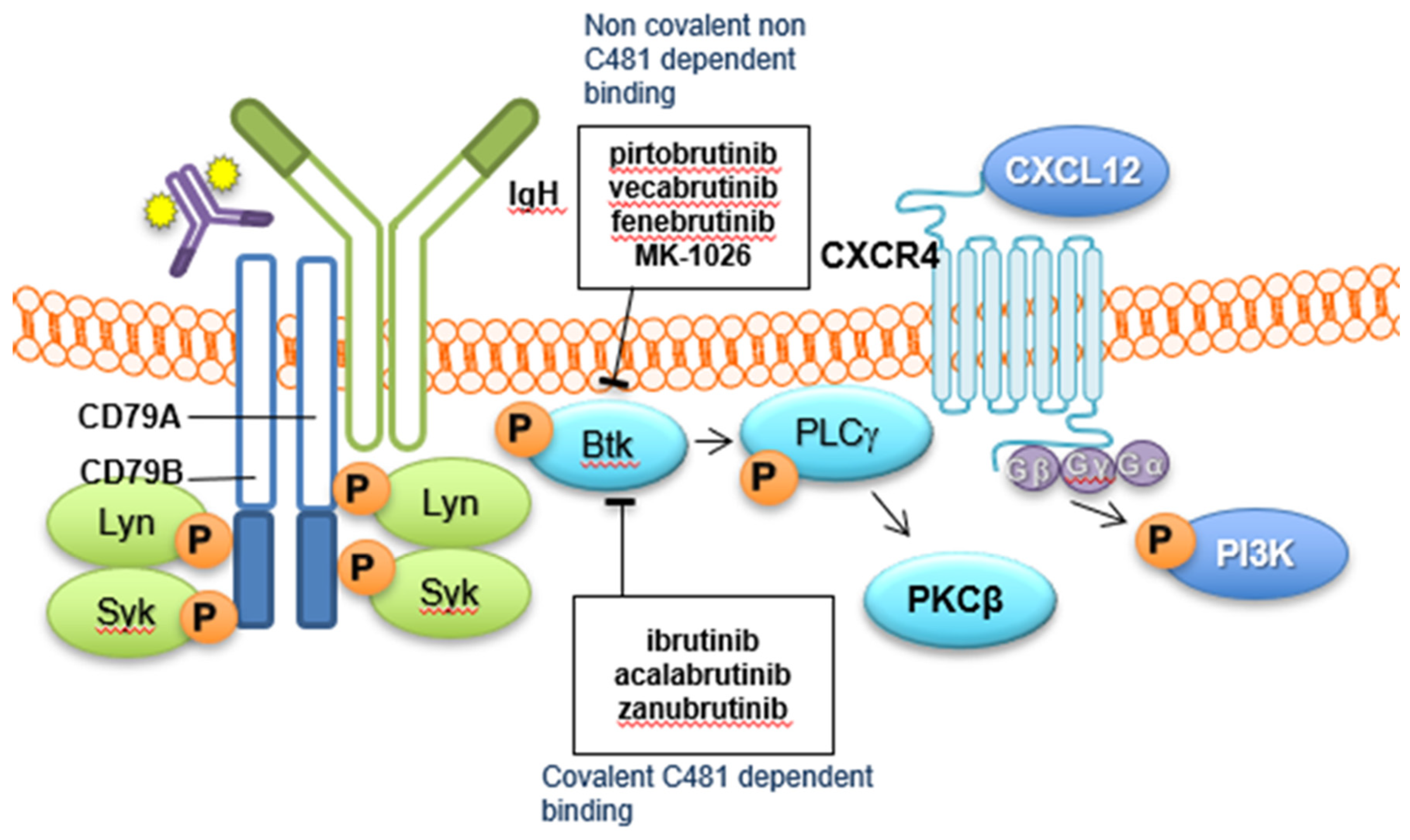
:1. Introduction
2. Covalent BTKi
2.1. Adverse Events with Covalent BTKi
2.2. Mechanisms of Resistance with Covalent BTKi
3. Non-Covalent BTKi—Clinical Development
3.1. Pirtobrutinib (LOXO-305)
3.2. Fenebrutinib (GDC-0853)
3.3. Vecabrutinib (SNS-062)
3.4. MK-1026
4. Non-Covalent BTKi—Preclinical Development
5. Combinations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Vetrie, D.; Vořechovský, I.; Sideras, P.; Holland, J.; Davies, A.; AFlinter, F.; Hammarström, L.; Kinnon, C.; Levinsky, R.J.; Bobrow, M.; et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nat. Cell Biol. 1993, 361, 226–233. [Google Scholar] [CrossRef]
- Tsukada, S.; Saffran, D.C.; Rawlings, D.J.; Parolini, O.; Allen, R.; Klisak, I.; Sparkes, R.S.; Kubagawa, H.; Mohandas, T.; Quan, S.; et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72, 279–290. [Google Scholar] [CrossRef]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef]
- Miao, Y.; Medeiros, L.J.; Li, Y.; Li, J.; Young, K.H. Genetic alterations and their clinical implications in DLBCL. Nat. Rev. Clin. Oncol. 2019, 16, 634–652. [Google Scholar] [CrossRef] [PubMed]
- Hershkovitz-Rokah, O.; Pulver, D.; Lenz, G.; Shpilberg, O. Ibrutinib resistance in mantle cell lymphoma: Clinical, molecular and treatment aspects. Br. J. Haematol. 2018, 181, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.N.R.; Bittner, Z.; Liu, X.; Dang, T.-M.; Radsak, M.P.; Brunner, C. Bruton’s Tyrosine Kinase: An Emerging Key Player in Innate Immunity. Front. Immunol. 2017, 8, 1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, M.; Treanor, B.; Depoil, D.; Shinohara, H.; Harwood, N.E.; Hikida, M.; Kurosaki, T.; Batista, F.D. Phospholipase C-gamma2 and Vav cooperate within signaling microclusters to propagate B cell spreading in response to membrane-bound antigen. J. Exp. Med. 2008, 205, 853–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, J.A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Tedeschi, A.; Bairey, O.; Hillmen, P.; Coutre, S.E.; Devereux, S.; et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia 2020, 34, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Sharman, J.P.; Egyed, M.; Jurczak, P.W.; Skarbnik, A.; Pagel, J.M.; Flinn, L.W.; Kamdar, M.; Munir, T.; Walewska, R.; Corbett, G.; et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): A randomised, controlled, phase 3 trial. Lancet 2020, 395, 1278–1291. [Google Scholar] [CrossRef]
- Tam, C.S.; Opat, S.; D’Sa, S.; Jurczak, W.; Lee, H.-P.; Cull, G.; Owen, R.G.; Marlton, P.; Wahlin, B.E.; Sanz, R.G.; et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: The ASPEN study. Blood 2020, 136, 2038–2050. [Google Scholar] [CrossRef]
- Xu, W.; Song, Y.; Li, Z.; Yang, S.; Liu, L.; Hu, Y.; Zhang, W.; Zhou, J.; Gao, S.; Ding, K.; et al. Safety, Tolerability and Efficacy of Orelabrutinib, Once a Day, to Treat Chinese Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia/Small Cell Leukemia. Blood 2019, 134, 4319. [Google Scholar] [CrossRef]
- Byrd, J.C.; Harrington, B.; O’Brien, S.; Jones, J.A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W.G.; Awan, F.T.; Brown, J.R.; et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Scheerens, H.; Li, S.-J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem Chem. Enabling Drug Discov. 2006, 2, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [Green Version]
- Advani, R.H.; Buggy, J.J.; Sharman, J.P.; Smith, S.M.; Boyd, T.E.; Grant, B.; Kolibaba, K.S.; Furman, R.R.; Rodriguez, S.; Chang, B.Y.; et al. Bruton Tyrosine Kinase Inhibitor Ibrutinib (PCI-32765) Has Significant Activity in Patients with Relapsed/Refractory B-Cell Malignancies. J. Clin. Oncol. 2013, 31, 88–94. [Google Scholar] [CrossRef]
- Dreyling, M.; Jurczak, W.; Jerkeman, M.; Silva, R.S.; Rusconi, C.; Trneny, M.; Offner, F.; Caballero, D.; João, C.; Witzens-Harig, M.; et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: An international, randomised, open-label, phase 3 study. Lancet 2016, 387, 770–778. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.; Greil, R.; Demirkan, F.; Tedeschi, A.; Anz, B.; Larratt, L.; Šimkovič, M.; Samoilova, O.; Novak, J.; Ben-Yehuda, D.; et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 43–56. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Wang, X.V.; Kay, N.E.; Hanson, C.A.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Ibrutinib–Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 432–443. [Google Scholar] [CrossRef]
- Woyach, J.A.; Ruppert, A.S.; Heerema, N.A.; Zhao, W.; Booth, A.M.; Ding, W.; Bartlett, N.; Brander, D.M.; Barr, P.M.; Rogers, K.A.; et al. Ibrutinib Regimens versus Chemoimmunotherapy in Older Patients with Untreated CLL. N. Engl. J. Med. 2018, 379, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N. Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Allan, J.N.; Shanafelt, T.; Wiestner, A.; Moreno, C.; O’Brien, S.M.; Braggio, E.; Liu, E.; Dean, J.P.; Lai, D.; Ahn, I.E. Long-Term Efficacy of First-Line Ibrutinib Treatment for Chronic Lymphocytic Leukemia (CLL) With 4 Years of Follow-Up in Patients with TP53 Aberrations (del (17p) or TP53 Mutation): A Pooled Analysis From 4 Clinical Trials. Blood 2020, 136, 23–24. [Google Scholar] [CrossRef]
- Munir, T.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Barr, P.M.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Final analysis from RESONATE: Up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am. J. Hematol. 2019, 94, 1353–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef]
- Jain, N.; Keating, M.; Thompson, P.; Ferrajoli, A.; Burger, J.; Borthakur, G.; Takahashi, K.; Estrov, Z.; Fowler, N.; Kadia, T.; et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N. Engl. J. Med. 2019, 380, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Tedeschi, A.; Trotman, J.; García-Sanz, R.; Macdonald, D.; Leblond, V.; Mahe, B.; Herbaux, C.; Tam, C.; Orsucci, L.; et al. Phase 3 Trial of Ibrutinib plus Rituximab in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2018, 378, 2399–2410. [Google Scholar] [CrossRef]
- Tam, C.S.; Ramchandren, R.; Chen, R.; Karlin, L.; Chong, G.; Jurczak, W.; Wu, K.L.; Bishton, M.M.; Collins, G.P.; Eliadis, M.P.; et al. Ibrutinib Plus Venetoclax in Patients with Relapsed/Refractory Mantle Cell Lymphoma: Results From the Safety Run-In Period of the Phase 3 Sympatico Study. Blood 2020, 136, 39–41. [Google Scholar] [CrossRef]
- Davids, M.S.; Brander, D.M.; Kim, H.T.; Tyekucheva, S.; Bsat, J.; Savell, A.; Hellman, J.M.; Bazemore, J.; Francoeur, K.; Alencar, A.; et al. Ibrutinib plus fludarabine, cyclophosphamide, and rituximab as initial treatment for younger patients with chronic lymphocytic leukaemia: A single-arm, multicentre, phase 2 trial. Lancet Haematol. 2019, 6, e419–e428. [Google Scholar] [CrossRef]
- Kater, A.; Owen, C.; Moreno, C.; Follows, G.; Munir, T.; Levin, M.-D.; Benjamini, O.; Janssens, A.; Osterborg, A.; Robak, T.; et al. Fixed-Duration Ibrutinib and Venetoclax (I+V) versus Chlorambucil Plus Obinutuzumab (CLB+O) for First-Line (1L) Chronic Lymphocytic Leukemia (CLL): Primary Analysis of the Phase 3 GLOW Study. Oral Abstract LB1902; EHA Virtual. 2021. Available online: https://library.ehaweb.org/eha/2021/eha2021-virtual-congress/330172/arnon.kater.fixed-duration.ibrutinib.and.venetoclax.28i2Bv29.versus.chlorambucil.html?f=menu%3D6%2Abrowseby%3D8%2Asortby%3D2%2Amedia%3D3%2Ace_id%3D2035%2Amarker%3D1284%2Afeatured%3D17286 (accessed on 5 March 2021).
- Harrington, B.K.; Gardner, H.L.; Izumi, R.; Hamdy, A.; Rothbaum, W.; Coombes, K.R.; Covey, T.; Kaptein, A.; Gulrajani, M.; Van Lith, B.; et al. Preclinical Evaluation of the Novel BTK Inhibitor Acalabrutinib in Canine Models of B-Cell Non-Hodgkin Lymphoma. PLoS ONE 2016, 11, e0159607. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, Y.; Hu, N.; Yu, D.; Zhou, C.; Shi, G.; Zhang, B.; Wei, M.; Liu, J.; Luo, L.; et al. Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase. J. Med. Chem. 2019, 62, 7923–7940. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, M.; Liu, D. Acalabrutinib (ACP-196): A selective second-generation BTK inhibitor. J. Hematol. Oncol. 2016, 9, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillmen, P.B.; Eichhorst, J.R. First interim analysis of ALPINE study: Results of a Phase 3 randomized study of Zanubrutinib vs Ibrutinib in patients with relapsed/refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. In Proceedings of the 25th European Hematology Association Annual Congress (EHA25), Virtual Meeting, 11–21 June 12020. [Google Scholar]
- Byrd, J.C.; Hillmen, P.; Ghia, P.; Kater, A.P.; Chanan-Khan, A.A.A.; Furman, R.R.; O’Brien, S.M.; Yenerel, M.N.; Illés, Á.; Kay, N.E.; et al. First Results of a head-to-head trial of Acalabrutinib versus Ibrutinib in previously treated Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2021, 39, 15. [Google Scholar] [CrossRef]
- Cheah, C.; Jurczak, W.; Lasica, M.; Wickham, N.W.; Wrobel, T.; Walewski, J.A.; Yannakou, C.K.; Cheung, S.; Lewis, K.L.; Długosz-Danecka, M.; et al. Updated results of the selective Bruton tyrosine kinase (BTK) inhibitor TG-1701, as monotherapy and in combination with ublituximab and umbralisib (U2) in patients (pts) with B-cell malignancies. J. Clin. Oncol. 2021, 39, 7525. [Google Scholar] [CrossRef]
- Narita, Y.; Nagane, M.; Mishima, K.; Terui, Y.; Arakawa, Y.; Yonezawa, H.; Asai, K.; Fukuhara, N.; Sugiyama, K.; Shinojima, N.; et al. Phase I/II study of tirabrutinib, a second-generation Bruton’s tyrosine kinase inhibitor, in relapsed/refractory primary central nervous system lymphoma. Neuro Oncol. 2021, 23, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Munakata, W.; Ando, K.; Hatake, K.; Fukuhara, N.; Kinoshita, T.; Fukuhara, S.; Shirasugi, Y.; Yokoyama, M.; Ichikawa, S.; Ohmachi, K.; et al. Phase I study of tirabrutinib (ONO-4059/GS-4059) in patients with relapsed or refractory B-cell malignancies in Japan. Cancer Sci. 2019, 110, 1686–1694. [Google Scholar] [CrossRef]
- Coutre, S.E.; Byrd, J.C.; Hillmen, P.; Barrientos, J.C.; Barr, P.M.; Devereux, S.; Robak, T.; Kipps, T.J.; Schuh, A.; Moreno, C.; et al. Long-term safety of single-agent ibrutinib in patients with chronic lymphocytic leukemia in 3 pivotal studies. Blood Adv. 2019, 3, 1799–1807. [Google Scholar] [CrossRef] [Green Version]
- Barr, P.M.; Robak, T.; Owen, C.; Tedeschi, A.; Bairey, O.; Bartlett, N.; Burger, J.A.; Hillmen, P.; Coutre, S.; Devereux, S.; et al. Sustained efficacy and detailed clinical follow-up of first-line ibrutinib treatment in older patients with chronic lymphocytic leukemia: Extended phase 3 results from RESONATE-2. Haematologica 2018, 103, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.M.; Jaglowski, S.; Byrd, J.C.; Bannerji, R.; Blum, K.A.; Fox, C.P.; Furman, R.R.; Hillmen, P.; Kipps, T.J.; Montillo, M.; et al. Prognostic Factors for Complete Response to Ibrutinib in Patients with Chronic Lymphocytic Leukemia: A Pooled Analysis of 2 Clinical Trials. JAMA Oncol. 2018, 4, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Estupiñán, H.Y.; Berglöf, A.; Zain, R.; Smith, C.I.E. Comparative Analysis of BTK Inhibitors and Mechanisms Underlying Adverse Effects. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Burger, J.A.; Blum, K.A.; Coleman, M.; Wierda, W.G.; Jones, J.A.; Zhao, W.; Heerema, N.A.; et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015, 125, 2497–2506. [Google Scholar] [CrossRef]
- Jain, P.; Keating, M.; Wierda, W.; Estrov, Z.; Ferrajoli, A.; Jain, N.; George, B.; James, D.; Kantarjian, H.; Burger, J.; et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood 2015, 125, 2062–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mato, A.R.; Nabhan, C.; Thompson, M.C.; Lamanna, N.; Brander, D.M.; Hill, B.; Howlett, C.; Skarbnik, A.; Cheson, B.D.; Zent, C.; et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: A real-world analysis. Haematologica 2018, 103, 874–879. [Google Scholar] [CrossRef]
- Yazdy, M.S.; Mato, A.R.; Roeker, L.E.; Jarral, U.; Ujjani, C.S.; Shadman, M.; Skarbnik, A.; Whitaker, K.J.; Deonarine, I.; Kabel, C.C.; et al. Toxicities and Outcomes of Acalabrutinib-Treated Patients with Chronic Lymphocytic Leukemia: A Retrospective Analysis of Real World Patients. Blood 2019, 134, 4311. [Google Scholar] [CrossRef]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.-H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [Green Version]
- Kaur, V.; Swami, A. Ibrutinib in CLL: A focus on adverse events, resistance, and novel approaches beyond ibrutinib. Ann. Hematol. 2017, 96, 1175–1184. [Google Scholar] [CrossRef]
- Puła, B.; Gołos, A.; Górniak, P.; Jamroziak, K. Overcoming Ibrutinib Resistance in Chronic Lymphocytic Leukemia. Cancers 2019, 11, 1834. [Google Scholar] [CrossRef] [Green Version]
- Woyach, J.A.; Johnson, A.J. Targeted therapies in CLL: Mechanisms of resistance and strategies for management. Blood 2015, 126, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.; Romaguera, J.E.; Williams, M.E.; et al. Targeting BTK with Ibrutinib in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.M.; Staudt, L.M. Ibrutinib Treatment of CLL: The Cancer Fights Back. Cancer Cell 2014, 26, 11–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddocks, K.J.; Ruppert, A.S.; Lozanski, G.; Heerema, N.A.; Zhao, W.; Abruzzo, L.V.; Lozanski, A.; Davis, M.; Gordon, A.L.; Smith, L.L.; et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients with Chronic Lymphocytic Leukemia. JAMA Oncol. 2015, 1, 80–87. [Google Scholar] [CrossRef]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTK(C481S)-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadri, S.; Lee, J.; Fitzpatrick, C.; Galanina, N.; Sukhanova, M.; Venkataraman, G.; Sharma, S.; Long, B.; Petras, K.; Theissen, M.; et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017, 1, 715–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Tsakmaklis, N.; Yang, G.; Chen, J.G.; Liu, X.; Demos, M.; Kofides, A.; Patterson, C.J.; Meid, K.; Gustine, J.; et al. Acquired mutations associated with ibrutinib resistance in Waldenström macroglobulinemia. Blood 2017, 129, 2519–2525. [Google Scholar] [CrossRef] [Green Version]
- Woyach, J.; Huang, Y.; Rogers, K.; Bhat, S.A.; Grever, M.R.; Lozanski, A.; Doong, T.-J.; Blachly, J.S.; Lozanski, G.; Jones, D.; et al. Resistance to Acalabrutinib in CLL is Mediated Primarily by BTK Mutations. Blood 2019, 134, 504. [Google Scholar] [CrossRef]
- Handunnetti, S.M.; Tang, C.P.S.; Nguyen, T.; Zhou, X.; Thompson, E.; Sun, H.; Xing, H.; Zhang, B.; Guo, Y.; Sutton, L.A.; et al. BTK Leu528Trp—A Potential Secondary Resistance Mechanism Specific for Patients with Chronic Lymphocytic Leukemia Treated with the Next Generation BTK Inhibitor Zanubrutinib. Blood 2019, 134 (Suppl. 1), 170. [Google Scholar] [CrossRef]
- Jain, P.; Kanagal-Shamanna, R.; Zhang, S.; Ahmed, M.; Ghorab, A.; Zhang, L.; Ok, C.Y.; Li, S.; Hagemeister, F.; Zeng, D.; et al. Long-term outcomes and mutation profiling of patients with mantle cell lymphoma (MCL) who discontinued ibrutinib. Br. J. Haematol. 2018, 183, 578–587. [Google Scholar] [CrossRef] [Green Version]
- Brandhuber, B.; Gomez, E.; Smith, S.; Eary, T.; Spencer, S.; Rothenberg, S.M.; Andrews, S. LOXO-305, A Next Generation Reversible BTK Inhibitor, for Overcoming Acquired Resistance to Irreversible BTK Inhibitors. Clin. Lymphoma Myeloma Leuk. 2018, 18, S216. [Google Scholar] [CrossRef]
- Mato, A.R.; Shah, N.N.; Jurczak, W.; Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.; Fakhri, B.; Eyre, T.A.; Lamanna, N.; Patel, M.R.; et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): A phase 1/2 study. Lancet 2021, 397, 892–901. [Google Scholar] [CrossRef]
- Mato, A.R.; Pagel, J.M.; Coombs, C.C.; Shah, N.N.; Lamanna, N.; Lech-Marańda, E.; Eyre, T.A.; Woyach, J.A.; Wierda, W.G.; Lewis, D.; et al. Pirtobrutinib (LOXO-305), a Next Generation, Highly Selective, Non-Covalent BTK inhibitor in previously treated Richter transformation: Results from the Phase 1/2 BRUIN study. Hematol. Oncol. 2021, 39, S2. [Google Scholar] [CrossRef]
- Byrd, J.C.; Smith, S.; Wagner-Johnston, N.; Sharman, J.; Chen, A.I.; Advani, R.; Augustson, B.; Marlton, P.; Commerford, S.R.; Okrah, K.; et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget 2018, 9, 13023–13035. [Google Scholar] [CrossRef] [Green Version]
- Fabian, C.A.; Reiff, S.D.; Guinn, D.; Neuman, L.; Fox, J.A.; Wilson, W.; Byrd, J.C.; Woyach, J.A.; Johnson, A.J. Abstract 1207: SNS-062 demonstrates efficacy in chronic lymphocytic leukemia in vitro and inhibits C481S mutated Bruton tyrosine kinase. Cancer Res. 2017, 77 (Suppl. 13), 1207. [Google Scholar]
- Neuman, M.L.L.; Ward, R.; Arnold, D.; Combs, B.D.L.; Gruver, M.D.; Hill, M.W.; Kunjom, J.M.; Miller, L.L.; Fox, J.A. First-in-Human Phase 1a Study of the Safety, Pharmacokinetics, and Pharmacodynamics of the Noncovalent Bruton Tyrosine Kinase (BTK) Inhibitor SNS-062 in Healthy Subjects. Blood 2016, 128, 2032. [Google Scholar] [CrossRef]
- Allan, J.N.; Patel, K.; Mato, A.R.; Wierda, W.G.; Pinilla Ibarz, J.; Choi, M.Y.; O’Brien, S.M.; Sharman, J.P.; Shadman, M.; Gladstone, D.E.; et al. Ongoing Results of a Phase 1B/2 Dose-Escalation and Cohort-Expansion Study of the Selective, Noncovalent, Reversible Bruton’S Tyrosine Kinase Inhibitor, Vecabrutinib, in B-Cell Malignancies. Blood 2019, 134, 3041. [Google Scholar] [CrossRef]
- Sunesis Pharmaceuticals, I. Sunesis Pharmaceuticals Announces Clinical Update on Vecabrutinib Program; Intrado GlobeNewswire: Omaha, NE, USA, 2020. [Google Scholar]
- Reiff, S.D.; Mantel, R.; Smith, L.L.; McWhorter, S.; Goettl, V.M.; Johnson, A.J.; Eathiraj, S.; Abbadessa, G.; Schwartz, B.; Byrd, J.C.; et al. The Bruton’s Tyrosine Kinase (BTK) Inhibitor ARQ 531 Effectively Inhibits Wild Type and C481S Mutant BTK and Is Superior to Ibrutinib in a Mouse Model of Chronic Lymphocytic Leukemia. Blood 2016, 128, 3232. [Google Scholar] [CrossRef]
- Woyach, J.; Stephens, D.D.M.; Flinn, I.W.; Bhat, S.A.; Savage, R.E.; Chai, F.; Eathiraj, S.; Granlund, L.; Szuszkiewicz, L.A.; Schwartz, B.; et al. Final Results of Phase 1, Dose Escalation Study Evaluating ARQ 531 in Patients with Relapsed or Refractory B-Cell Lymphoid Malignancies. Blood 2019, 134, 4298. [Google Scholar] [CrossRef]
- Gui, F.; Jiang, J.; He, Z.; Li, L.; Li, Y.; Deng, Z.; Lu, Y.; Wu, X.; Chen, G.; Su, J.; et al. A non-covalent inhibitor XMU-MP-3 overrides ibrutinib-resistant Btk(C481S) mutation in B-cell malignancies. Br. J. Pharmacol. 2019, 176, 4491–4509. [Google Scholar] [CrossRef] [PubMed]
- Asami, T.; Kawahata, W.; Kashimoto, S.; Sawa, M. Abstract B152: CB1763, a highly selective, novel non-covalent BTK inhibitor, targeting ibrutinib-resistant BTK C481S mutant. Mol. Cancer Ther. 2018, 17, B152. [Google Scholar] [CrossRef]
- Johnson, A.R.; Kohli, P.B.; Katewa, A.; Gogol, E.; Belmont, L.D.; Choy, R.; Penuel, E.; Burton, L.; Eigenbrot, C.; Yu, C.; et al. Battling Btk Mutants with Noncovalent Inhibitors That Overcome Cys481 and Thr474 Mutations. ACS Chem. Biol. 2016, 11, 2897–2907. [Google Scholar] [CrossRef]
- Guo, X.; Yang, D.; Fan, Z.; Zhang, N.; Zhao, B.; Huang, C.; Wang, F.; Ma, R.; Meng, M.; Deng, Y. Discovery and structure-activity relationship of novel diphenylthiazole derivatives as BTK inhibitor with potent activity against B cell lymphoma cell lines. Eur. J. Med. Chem. 2019, 178, 767–781. [Google Scholar] [CrossRef]


{kind=link}
| Non-Covalent BTKi | Pirtobrutinib | Fenebrutinib | Vecabrutinib | MK-1026 |
|---|---|---|---|---|
| Structure |  |  |  |  |
| Other names | LOXO-305 | GDC-0853 | SNS-062 | ARQ-531 |
| Binding to BTK | Blocks ATP binding site of BTK | Hydrogen bonds with K430, M477, D539 | Decreases surface expression of B-cell activation markers | Hydrogen bonds with E475, Y476 |
| Other enzyme activity | Minimal | Minimal | Activity on ITK No activity on EGFR | Activity on SRC, ERK, AKT Inhibits signalling downstream of PCLG2 |
| Side effects (%) | Fatigue (20%) Diarrhea (17%) Contusion (13%) Neutropenia (13%) | Fatigue (37.5%) Nausea (33%) Diarrhea (29%) Thrombocytopenia (25%) Headache (21%) | Anemia (37.5%) Headache (25%) Neutropenia (25%) Night sweats (25%) | Nausea (10%) Diarrhea (10%) Fatigue (8%) Neutropenia (8%) Dysgeusia (8%) Rash (8%) |
| Clinical development | Phase Ib/II ongoing in B-cell malignancies Phase III ongoing in MCL Phase III ongoing in CLL | Terminated in B-cell malignancies Ongoing in multiple sclerosis | Terminated in B-cell malignancies | Phase Ib ongoing in B-cell malignancies Phase II ongoing in B-cell malignancies |
| Key publications | [51,52] | [53] | [5,54,55] | [57,58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, K.L.; Cheah, C.Y. Non-Covalent BTK Inhibitors—The New BTKids on the Block for B-Cell Malignancies. J. Pers. Med. 2021, 11, 764. https://doi.org/10.3390/jpm11080764
Lewis KL, Cheah CY. Non-Covalent BTK Inhibitors—The New BTKids on the Block for B-Cell Malignancies. Journal of Personalized Medicine. 2021; 11(8):764. https://doi.org/10.3390/jpm11080764
Chicago/Turabian StyleLewis, Katharine L, and Chan Y Cheah. 2021. "Non-Covalent BTK Inhibitors—The New BTKids on the Block for B-Cell Malignancies" Journal of Personalized Medicine 11, no. 8: 764. https://doi.org/10.3390/jpm11080764