
COVID-19 and Venous Thromboembolism: From Pathological Mechanisms to Clinical Management

Abstract
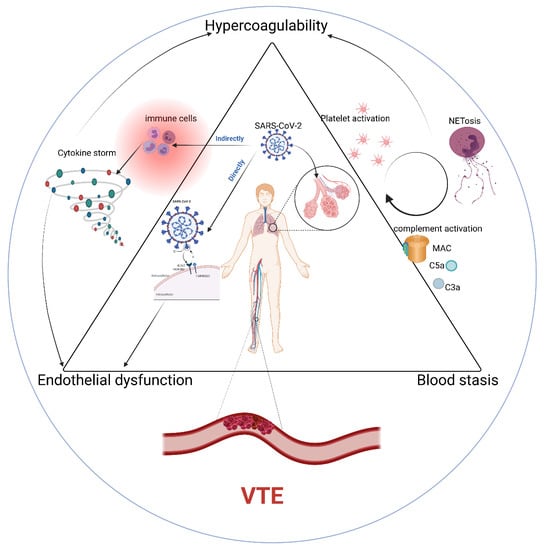
:
1. Introduction
2. Pathophysiology of VTE in COVID-19
Endothelial Dysfunction
3. Hypercoagulable State
3.1. Platelet Activation
3.2. Cytokines and Chemokines
3.3. Complement
3.4. Neutrophil Extracellular Traps/NETs
4. Abnormalities of Blood Flow
5. VTE Clinical Management
5.1. Anticoagulation Management
5.2. Antiplatelet Agents
5.3. Other Clinical Management
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.C.; et al. China Medical Treatment Expert Group for COVID-Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Wichmann, D.; Sperhake, J.P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.B.; Garcia, D.; Crowther, M.; Savage, B.; Peress, S.; Chang, K.; Deitelzweig, S. Frequency of venous thromboembolism in 6513 patients with COVID-19: A retrospective study. Blood Adv. 2020, 4, 5373–5377. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, D.; García-Sanchez, A.; Rali, P.; Muriel, A.; Bikdeli, B.; Ruiz-Artacho, P.; Le Mao, R.; Rodríguez, C.; Hunt, B.J.; Monreal, M. Incidence of VTE and Bleeding Among Hospitalized Patients with Coronavirus Disease 2019: A Systematic Review and Meta-analysis. Chest 2021, 159, 1182–1196. [Google Scholar] [CrossRef] [PubMed]
- Lax, S.F.; Skok, K.; Zechner, P.; Kessler, H.H.; Kaufmann, N.; Koelblinger, C.; Vander, K.; Bargfrieder, U.; Trauner, M. Pulmonary Arterial Thrombosis in COVID-19 With Fatal Outcome: Results from a Prospective, Single-Center, Clinicopathologic Case Series. Ann. Intern. Med. 2020, 173, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Anderson, F.A., Jr.; Spencer, F.A. Risk factors for venous thromboembolism. Circulation 2003, 107 (Suppl. 1), I-9–I-16. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Zuo, Z.; Yang, D.; Luo, X.; Jiang, L.; Xia, Z.; Xiao, X.; Liu, J.; Ye, M.; Deng, M. Venous thromboembolic events in patients with COVID-19: A systematic review and meta-analysis. Age Ageing 2021, 50, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Gianos, E.; Barish, M.A.; Chatterjee, S.; Kohn, N.; Lesser, M.; Giannis, D.; Coppa, K.; Hirsch, J.; McGinn, T.; et al. Prevalence and Predictors of Venous Thromboembolism or Mortality in Hospitalized COVID-19 Patients. Thromb. Haemost. 2021, 121, 1043–1053. [Google Scholar] [CrossRef]
- Mehta, J.L.; Calcaterra, G.; Bassareo, P.P. COVID-19, thromboembolic risk, and Virchow’s triad: Lesson from the past. Clin. Cardiol. 2020, 43, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Lüscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arter. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef]
- Hadi, H.A.R.; Carr, C.S.; Al Suwaidi, J. Endothelial Dysfunction: Cardiovascular Risk Factors, Therapy, and Outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; Macary, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Teuwen, L.-A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Abassi, Z.; Skorecki, K.; Hamo-Giladi, D.B.; Kruzel-Davila, E.; Heyman, S.N. Kinins and chymase: The forgotten components of the renin-angiotensin system and their implications in COVID-19 disease. Am. J. Physiol. Cell. Mol. Physiol. 2021, 320, L422–L429. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Mendes, M.C.; Martins, A.P.C.; Borges, N.H.; Godoy, T.M.; Dos Santos Miggiolaro, A.F.R.; Dos Santos Dezidério, F.; Machado-Souza, C.; de Noronha, L. Endothelial Dysfunction and Thrombosis in Patients with COVID-19—Brief Report. Arter. Thromb. Vasc. Biol. 2020, 40, 2404–2407. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zou, M.-H. Molecular Insights and Therapeutic Targets for Diabetic Endothelial Dysfunction. Circulation 2009, 120, 1266–1286. [Google Scholar] [CrossRef] [Green Version]
- Loo, J.; Spittle, D.A.; Newnham, M. COVID-19, immunothrombosis and venous thromboembolism: Biological mechanisms. Thorax 2021, 76, 412–420. [Google Scholar] [CrossRef]
- Marchetti, M. COVID-19-driven endothelial damage: Complement, HIF-1, and ABL2 are potential pathways of damage and targets for cure. Ann. Hematol. 2020, 99, 1701–1707. [Google Scholar] [CrossRef]
- Stevens, H.; McFadyen, J.D. Platelets as Central Actors in Thrombosis—Reprising an Old Role and Defining a New Character. Semin. Thromb. Hemost. 2019, 45, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Ahmad, F.; Kannan, M.; Ansari, A.W. Role of SARS-CoV-2 -induced cytokines and growth factors in coagulopathy and thromboembolism. Cytokine Growth Factor Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Stitham, J.; Jin, Y.; Liu, R.; Lee, S.H.; Du, J.; Atteya, G.; Gleim, S.; Spollett, G.; Martin, K.; et al. Aldose Reductase–Mediated Phosphorylation of p53 Leads to Mitochondrial Dysfunction and Damage in Diabetic Platelets. Circulation 2014, 129, 1598–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Gu, S.X.; Tyagi, T.; Jain, K.; Gu, V.W.; Lee, S.H.; Hwa, J.M.; Kwan, J.M.; Krause, D.S.; Lee, A.I.; Halene, S.; et al. Thrombocytopathy and endotheliopathy: Crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 2021, 18, 194–209. [Google Scholar] [CrossRef]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F.; et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg. Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Du, F.; Liu, B.; Zhang, S. COVID-19: The role of excessive cytokine release and potential ACE2 down-regulation in promoting hypercoagulable state associated with severe illness. J. Thromb. Thrombolysis 2021, 51, 313–329. [Google Scholar] [CrossRef]
- Bertin, F.; Rys, R.N.; Mathieu, C.; Laurance, S.; Lemarié, C.A.; Blostein, M.D. Natural killer cells induce neutrophil extracellular trap formation in venous thrombosis. J. Thromb. Haemost. 2019, 17, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168. [Google Scholar] [CrossRef]
- Gao, H.; Zhang, Q.; Chen, J.; Cooper, D.K.; Hara, H.; Chen, P.; Wei, L.; Zhao, Y.; Xu, J.; Li, Z.; et al. Porcine IL-6, IL-1β, and TNF-α regulate the expression of pro-inflammatory-related genes and tissue factor in human umbilical vein endothelial cells. Xenotransplantation 2018, 25, e12408. [Google Scholar] [CrossRef]
- Baars, J.W.; de Boer, J.P.; Wagstaff, J.; Roem, D.; Eerenberg-Belmer, A.J.M.; Nauta, J.; Pinedo, H.M.; Hack, C.E. Interleukin-2 induces activation of coagulation and fibrinolysis: Resemblance to the changes seen during experimental endotoxaemia. Br. J. Haematol. 1992, 82, 295–301. [Google Scholar] [CrossRef]
- Wojta, J.; Huber, K.; Valent, P. New Aspects in Thrombotic Research: Complement Induced Switch in Mast Cells from a Profibrinolytic to a Prothrombotic Phenotype. Pathophysiol. Haemost. Thromb. 2003, 33, 438–441. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor–enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef] [PubMed]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 2020, 75, 1564–1581. [Google Scholar] [CrossRef] [PubMed]
- Fletcher-Sandersjöö, A.; Bellander, B.M. Is COVID-19 associated thrombosis caused by overactivation of the complement cascade? A literature review. Thromb. Res. 2020, 194, 36–41. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Brown, J.Q.; Heide, R.S.V. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-J.; Jenne, C.N. Role of platelets in neutrophil extracellular trap (NET) production and tissue injury. Semin. Immunol. 2016, 28, 546–554. [Google Scholar] [CrossRef]
- Gollomp, K.; Kim, M.; Johnston, I.; Hayes, V.; Welsh, J.; Arepally, G.M.; Kahn, M.; Lambert, M.P.; Cuker, A.; Cines, D.B.; et al. Neutrophil accumulation and NET release contribute to thrombosis in HIT. JCI Insight 2018, 3, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Furie, B.C. The biology of P-selectin glycoprotein ligand-1: Its role as a selectin counterreceptor in leukocyte-endothelial and leukocyte-platelet interaction. Thromb. Haemost. 1999, 81, 1–7. [Google Scholar]
- Faraday, N.; Schunke, K.; Saleem, S.; Fu, J.; Wang, B.; Zhang, J.; Morrell, C.; Doré, S. Cathepsin G-Dependent Modulation of Platelet Thrombus Formation In Vivo by Blood Neutrophils. PLoS ONE 2013, 8, e71447. [Google Scholar] [CrossRef] [Green Version]
- Goel, M.S.; Diamond, S.L. Neutrophil cathepsin G promotes prothrombinase and fibrin formation under flow conditions by activating fibrinogen-adherent platelets. J. Biol. Chem. 2003, 278, 9458–9463. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Peng, R.; Wang, F.; Hua, L.; Liu, S.; Han, Z.; Pei, J.; Pei, S.; Zhao, Z.; Jiang, X.; et al. Lysophosphatidic acid promotes thrombus stability by inducing rapid formation of neutrophil extracellular traps: A new mechanism of thrombosis. J. Thromb. Haemost. 2020, 18, 1952–1964. [Google Scholar] [CrossRef]
- Savchenko, A.S.; Martinod, K.; Seidman, M.; Wong, S.L.; Borissoff, J.I.; Piazza, G.; Libby, P.; Goldhaber, S.Z.; Mitchell, R.N.; Wagner, D.D. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J. Thromb. Haemost. 2014, 12, 860–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.D.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef]
- Edler, C.; Schröder, A.S.; Aepfelbacher, M.; Fitzek, A.; Heinemann, A.; Heinrich, F.; Klein, A.; Langenwalder, F.; Lütgehetmann, M.; Meißner, K.; et al. Dying with SARS-CoV-2 infection-an autopsy study of the first consecutive 80 cases in Hamburg, Germany. Int. J. Legal Med. 2020, 134, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulos, A.C.; Levy, J.H.; Ageno, W.; Connors, J.M.; Hunt, B.J.; Iba, T.; Levi, M.; Samama, C.M.; Thachil, J.; Giannis, D.; et al. Scientific and Standardization Committee communication: Clinical guidance on the diagnosis, prevention, and treatment of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Poterucha, T.J.; Libby, P.; Goldhaber, S.Z. More than an anticoagulant: Do heparins have direct anti-inflammatory effects? Thromb. Haemost. 2017, 117, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.D.; Moore, H.B.; Yaffe, M.B.; Moore, E.E. ISTH interim guidance on recognition and management of coagulopathy in COVID-19: A comment. J. Thromb. Haemost. 2020, 18, 2060–2063. [Google Scholar] [CrossRef] [PubMed]
- Thachil, J. The versatile heparin in COVID-19. J. Thromb. Haemost. 2020, 18, 1020–1022. [Google Scholar] [CrossRef] [Green Version]
- Warkentin, T.E. Laboratory diagnosis of heparin-induced thrombocytopenia. Int. J. Lab. Hematol. 2019, 41 (Suppl. 1), 15–25. [Google Scholar] [CrossRef] [Green Version]
- Paranjpe, I.; Fuster, V.; Lala, A.; Russak, A.J.; Glicksberg, B.S.; Levin, M.A.; Charney, A.W.; Narula, J.; Fayad, Z.A.; Bagiella, E.; et al. Association of Treatment Dose Anticoagulation with In-Hospital Survival Among Hospitalized Patients with COVID-19. J. Am. Coll. Cardiol. 2020, 76, 122–124. [Google Scholar] [CrossRef]
- Testa, S.; Paoletti, O.; Giorgi-Pierfranceschi, M.; Pan, A. Switch from oral anticoagulants to parenteral heparin in SARS-CoV-2 hospitalized patients. Intern. Emerg. Med. 2020, 15, 751–753. [Google Scholar] [CrossRef] [Green Version]
- Testa, S.; Prandoni, P.; Paoletti, O.; Morandini, R.; Tala, M.; Dellanoce, C.; Giorgi-Pierfranceschi, M.; Betti, M.; Danzi, G.B.; Pan, A.; et al. Direct oral anticoagulant plasma levels’ striking increase in severe COVID-19 respiratory syndrome patients treated with antiviral agents: The Cremona experience. J. Thromb. Haemost. 2020, 18, 1320–1323. [Google Scholar] [CrossRef]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. DOACs and “newer” hemophilia therapies in COVID-19: Reply. J. Thromb. Haemost. 2020, 18, 1795–1796. [Google Scholar] [CrossRef]
- Viecca, M.; Radovanovic, D.; Forleo, G.B.; Santus, P. Enhanced platelet inhibition treatment improves hypoxemia in patients with severe COVID-19 and hypercoagulability. A case control, proof of concept study. Pharmacol. Res. 2020, 158, 104950. [Google Scholar] [CrossRef]
- Liu, X.; Li, Z.; Liu, S.; Sun, J.; Chen, Z.; Jiang, M.; Zhang, Q.; Wei, Y.; Wang, X.; Huang, Y.-Y.; et al. Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm. Sin. B 2020, 10, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Asakura, H.; Ogawa, H. Potential of heparin and nafamostat combination therapy for COVID-19. J. Thromb. Haemost. 2020, 18, 1521–1522. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Ino, Y.; Ozeki, M.; Oda, M.; Sato, T.; Koshiyama, Y.; Suzuki, S.; Fujita, M. Pharmacological Studies of FUT-175, Nafamstat Mesilate I. Inhibition of Protease Activity in In Vitro and In Vivo Experiments. Jpn. J. Pharmacol. 1984, 35, 203–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, Y.; Yoshida, H.; Uchino, S.; Yokoyama, K.; Yamamoto, H.; Takinami, M.; Hosoya, T. Nafamostat Mesilate as an Anticoagulant during Continuous Veno-Venous Hemodialysis: A Three-Year Retrospective Cohort Study. Int. J. Artif. Organs 2011, 34, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Karakas, M.; Jarczak, D.; Becker, M.; Roedl, K.; Addo, M.M.; Hein, F.; Bergmann, A.; Zimmermann, J.; Simon, T.P.; Marx, G.; et al. Targeting Endothelial Dysfunction in Eight Extreme-Critically Ill Patients with COVID-19 Using the Anti-Adrenomedullin Antibody Adrecizumab (HAM8101). Biomolecules 2020, 10, 1171. [Google Scholar] [CrossRef]
- Hammarén, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef]
- Masiá, M.; Fernández-González, M.; Padilla, S.; Ortega, P.; García, J.A.; Agulló, V.; García-Abellán, J.; Telenti, G.; Guillén, L.; Gutiérrez, F. Impact of interleukin-6 blockade with tocilizumab on SARS-CoV-2 viral kinetics and antibody responses in patients with COVID-19: A prospective cohort study. EBioMedicine 2020, 60, 102999. [Google Scholar] [CrossRef]
- Maslennikov, R.; Ivashkin, V.; Vasilieva, E.; Chipurik, M.; Semikova, P.; Semenets, V.; Russkova, T.; Levshina, A.; Grigoriadis, D.; Magomedov, S.; et al. Tofacitinib reduces mortality in coronavirus disease 2019 Tofacitinib in COVID-19. Pulm. Pharmacol. Ther. 2021, 69, 102039. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Wright, F.; Vogler, T.O.; Moore, E.E.; Moore, H.B.; Wohlauer, M.V.; Urban, S.; Nydam, T.L.; Moore, P.K.; McIntyre, R.C., Jr. Fibrinolysis Shutdown Correlation with Thromboembolic Events in Severe COVID-19 Infection. J. Am. Coll. Surg. 2020, 231, 193–203. [Google Scholar] [CrossRef]
- Whyte, C.S.; Morrow, G.B.; Mitchell, J.L.; Chowdary, P.; Mutch, N.J. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombo-lytic drugs to treat COVID-19. J. Thromb. Haemost. 2020, 18, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and Antiphospholipid Antibodies in Patients with COVID-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef] [PubMed]
- Diurno, F.; Numis, F.G.; Porta, G.; Cirillo, F.; Maddaluno, S.; Ragozzino, A.; De Negri, P.; Di Gennaro, C.; Pagano, A.; Allegorico, E.; et al. Eculizumab treatment in patients with COVID-19: Preliminary results from real life ASL Napoli 2 Nord experience. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4040–4047. [Google Scholar] [PubMed]
- McEneny-King, A.C.; Monteleone, J.P.R.; Kazani, S.D.; Ortiz, S.R. Pharmacokinetic and Pharmacodynamic Evaluation of Ravulizumab in Adults with Severe Coronavirus Disease 2019. Infect. Dis. Ther. 2021, 10, 1045–1054. [Google Scholar] [CrossRef]

{kind=link}
| ClinicalTrials.gov Identifier | Study Design/Status | Patients, No. | Treatment Group |
|---|---|---|---|
| NCT04842292 | Interventional/Recruiting | 40 | Heparin vs. Placebo |
| NCT04746339 | Interventional/Recruiting | 1000 | Apixaban 2.5 mg vs. Placebo |
| NCT04650087 | Interventional/Recruiting | 5320 | Apixaban 2.5 mg vs. Placebo |
| NCT04600141 | Interventional/Recruiting | 308 | Tocilizumab vs. Heparin Therapeutic dosage vs. Heparin Prophylactic dosage |
| NCT04581954 | Interventional/Recruiting | 456 | Ruxolitinib vs. Fostamatinib vs. Standard of care |
| NCT04542408 | Interventional/Recruiting | 172 | Anticoagulation Agents (Edoxaban and/or high dose LMWH) vs. Low dose Low molecular weight heparin or Placebo |
| NCT04508023 | Interventional/Recruiting | 4000 | Rivaroxaban vs. Placebo vs. Standard of Care (SOC) |
| NCT04492254 | Interventional/Recruiting | 1370 | Enoxaparin |
| NCT04486508 | Interventional/completed | 600 | Intermediate dose Enoxaparin/unfractionated heparin vs. Standard prophylactic dose Enoxaparin/unfractionated heparin vs. Atorvastatin 20 mg vs. Matched placebo |
| NCT04466670 | Interventional/Recruiting | 379 | Acetylsalicylic acid vs. Unfractionated heparin nebulized |
| NCT04416048 | Interventional/Recruiting | 400 | Rivaroxaban vs. Standard Of Care (SOC) |
| NCT04406389 | Interventional/Recruiting | 186 | Enoxaparin sodium vs. Unfractionated heparin vs. Fondapariniux vs. Argatroban |
| NCT04401293 | Interventional/completed | 257 | Enoxaparin vs. Prophylactic/Intermediate Dose Enoxaparin |
| NCT04394377 | Interventional/completed | 615 | Rivaroxaban20 mg/d followed by enoxaparin/unfractionated heparin when needed vs. Control group with enoxaparin 40mg/d |
| NCT04394000 | Interventional/completed | 72 | Thromboprofylaxis protocol vs. Standard protocol |
| NCT04373707 | Interventional/Recruiting | 602 | Enoxaparin |
| NCT04372589 | Interventional/completed | 1200 | Heparin |
| NCT04367831 | Interventional/Recruiting | 100 | Enoxaparin Prophylactic Dose vs. Heparin Infusion vs. Heparin SC vs. Enoxaparin/Lovenox Intermediate Dose |
| NCT04366960 | Interventional/completed | 189 | Enoxaparin |
| NCT04360824 | Interventional/Recruiting | 170 | Intermediate dose thromboprophylaxis vs. Standard of Care thromboprophylaxis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Cheng, Z.; Hu, Y. COVID-19 and Venous Thromboembolism: From Pathological Mechanisms to Clinical Management. J. Pers. Med. 2021, 11, 1328. https://doi.org/10.3390/jpm11121328
Zhou X, Cheng Z, Hu Y. COVID-19 and Venous Thromboembolism: From Pathological Mechanisms to Clinical Management. Journal of Personalized Medicine. 2021; 11(12):1328. https://doi.org/10.3390/jpm11121328
Chicago/Turabian StyleZhou, Xianghui, Zhipeng Cheng, and Yu Hu. 2021. "COVID-19 and Venous Thromboembolism: From Pathological Mechanisms to Clinical Management" Journal of Personalized Medicine 11, no. 12: 1328. https://doi.org/10.3390/jpm11121328