
Ace2 and Tmprss2 Expressions Are Regulated by Dhx32 and Influence the Gastrointestinal Symptoms Caused by SARS-CoV-2

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Tissue Collection
2.2. RNA Isolation and Transcriptome Data Generation
2.3. Data Preprocessing
2.4. Expression Data and FAIR Data Access
2.5. Expression Quantitative Trait Locus (eQTL) Mapping
2.6. Microbiome Analysis and Data Access
2.7. Correlation Analysis
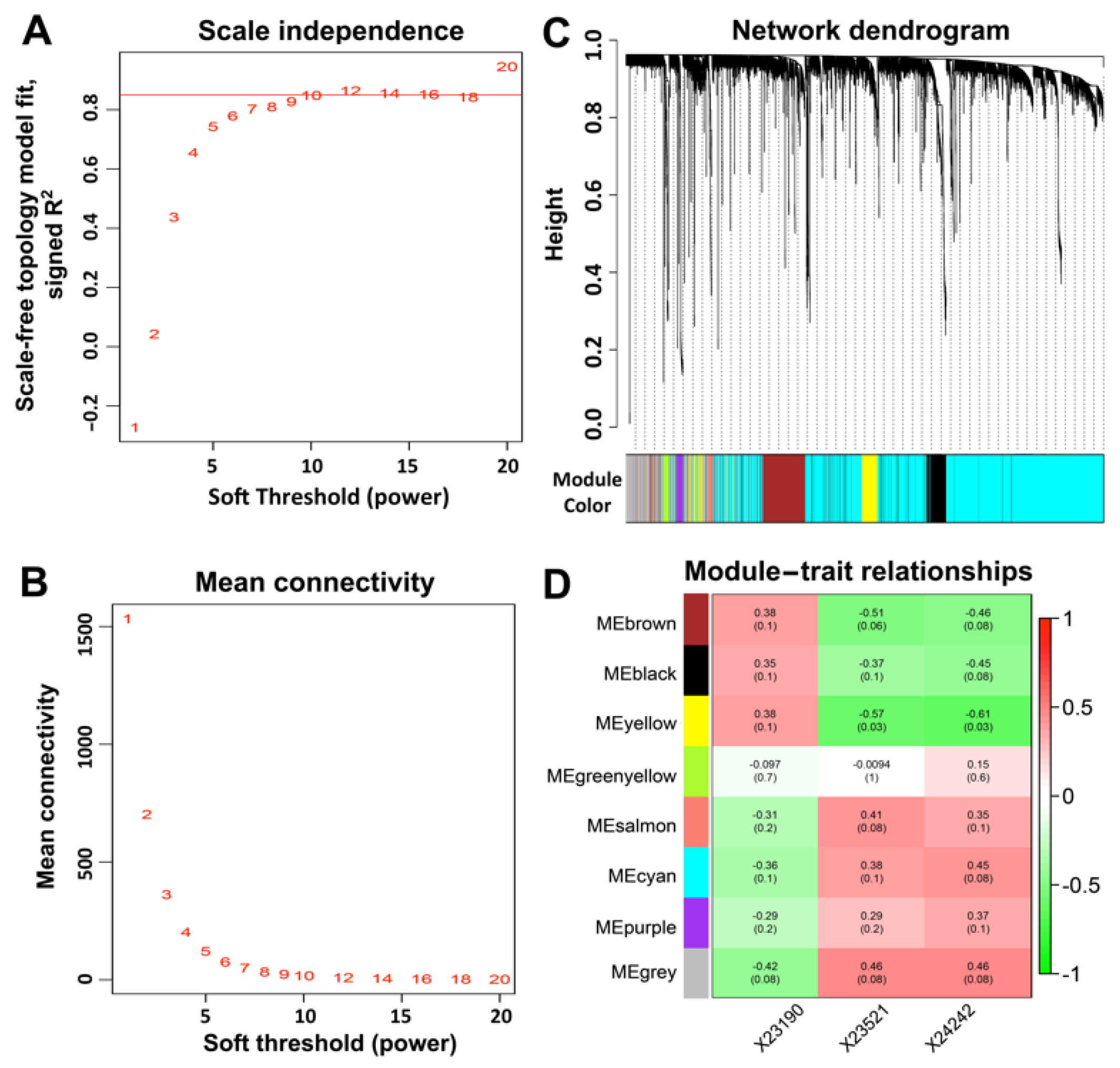
2.8. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.9. Gene Set Enrichment Analysis
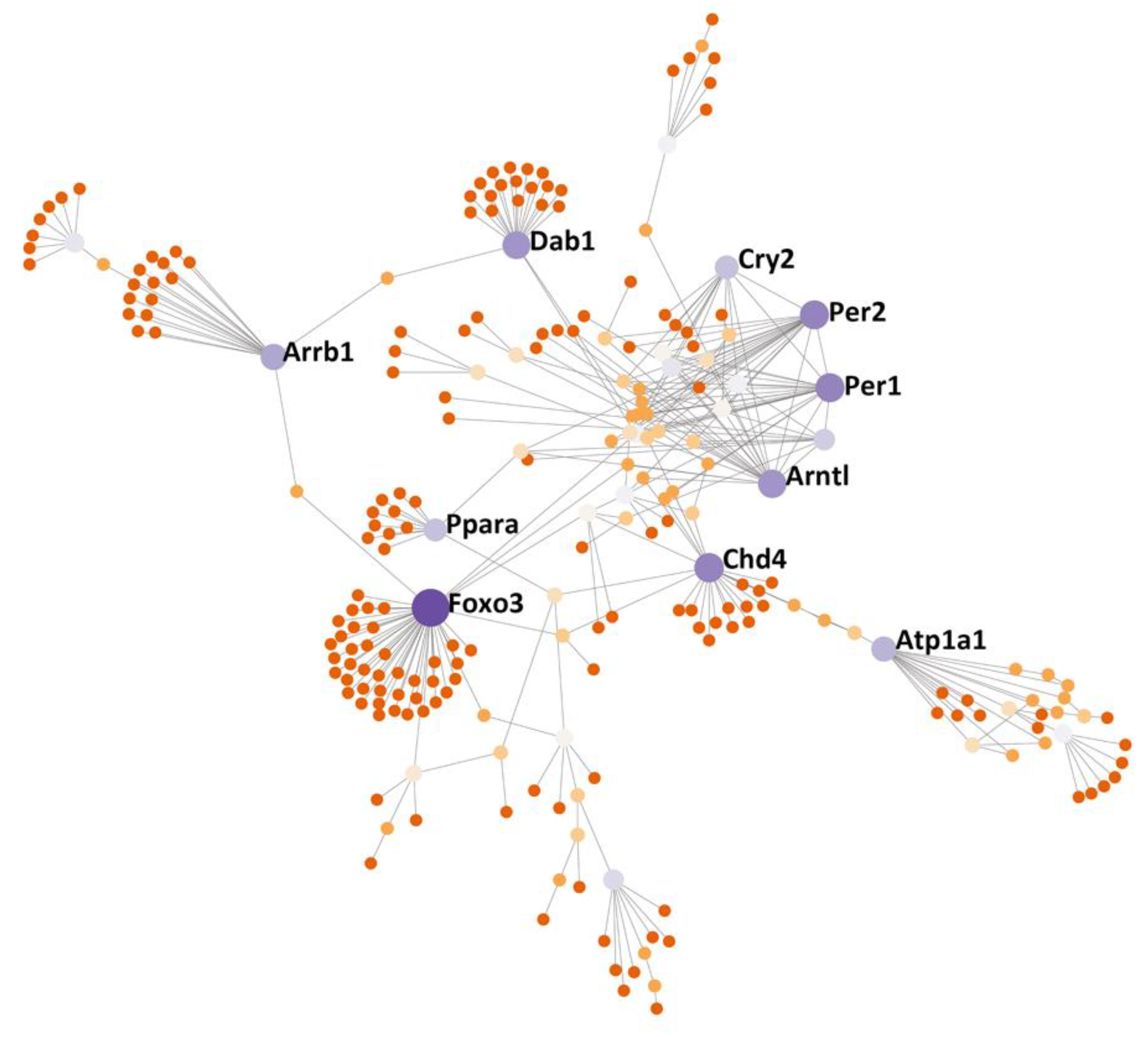
2.10. Protein–Protein Interactions (PPI) Analysis
3. Results
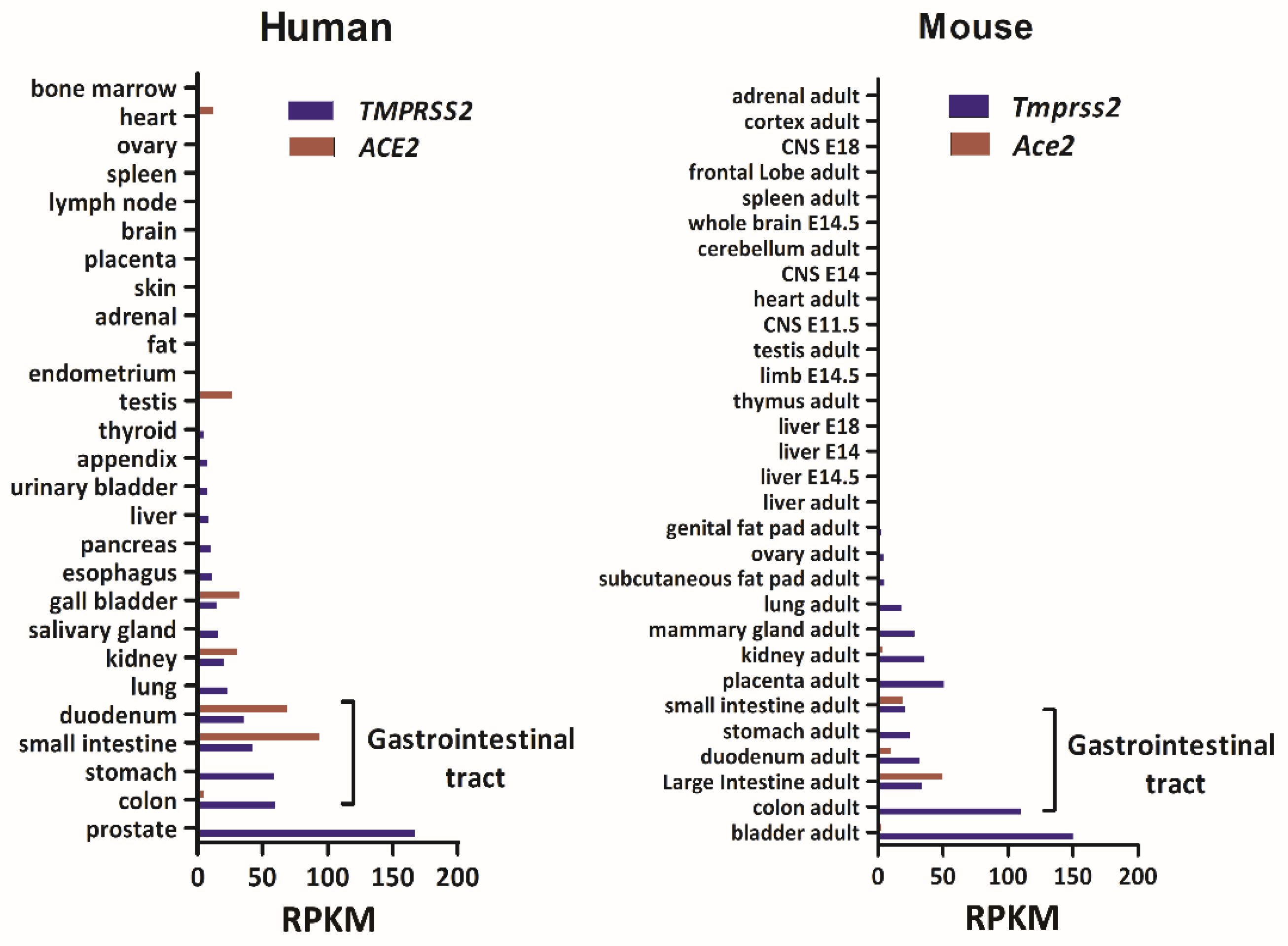
3.1. Tmprss2 and Ace2 mRNA Levels in Human and Mouse Tissues
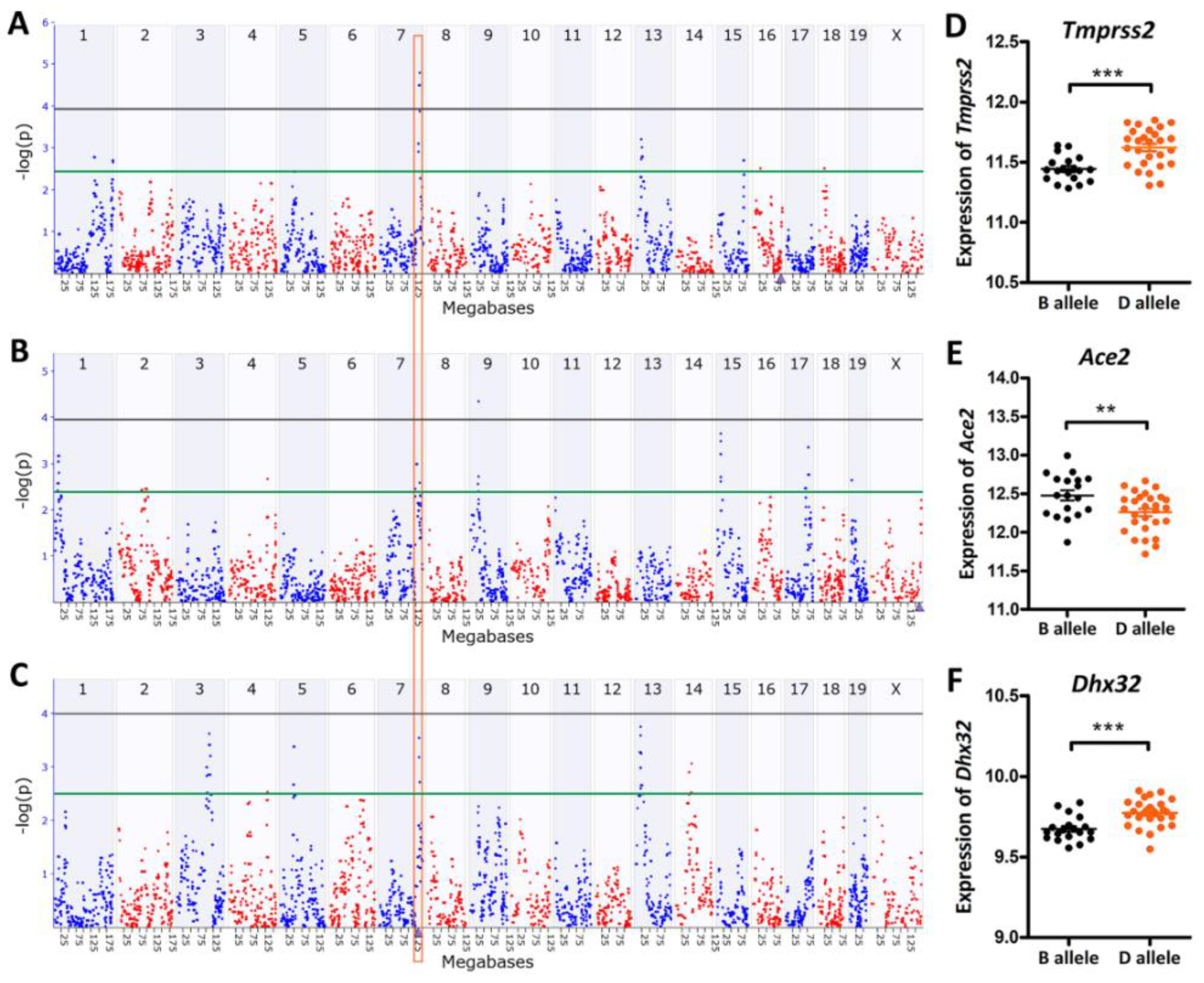
3.2. eQTL Mapping Identified a Common Regulating Locus for Tmprss2 and Ace2
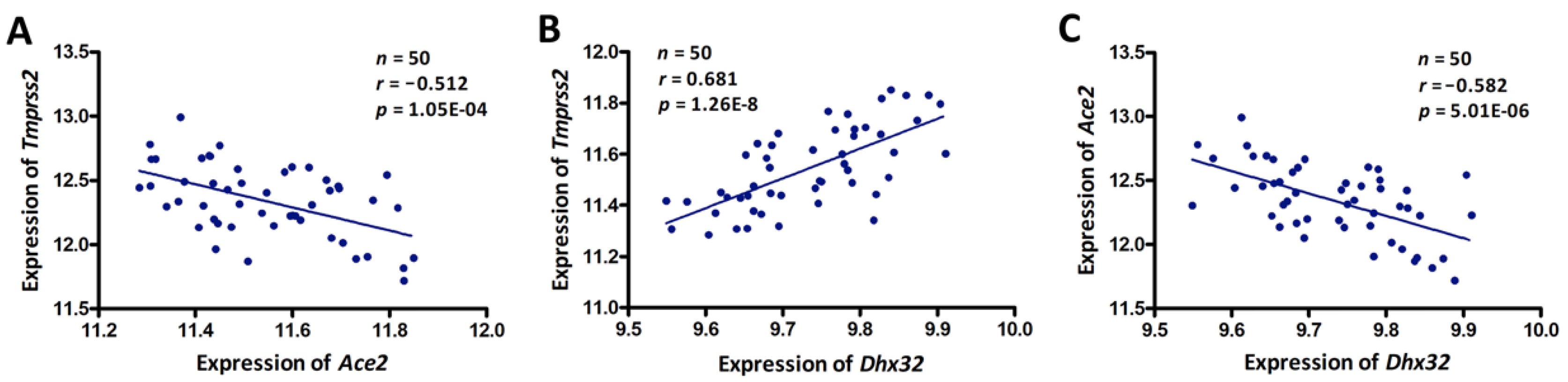
3.3. Dhx32 was a Candidate Upstream Regulator for Tmprss2 and Ace2
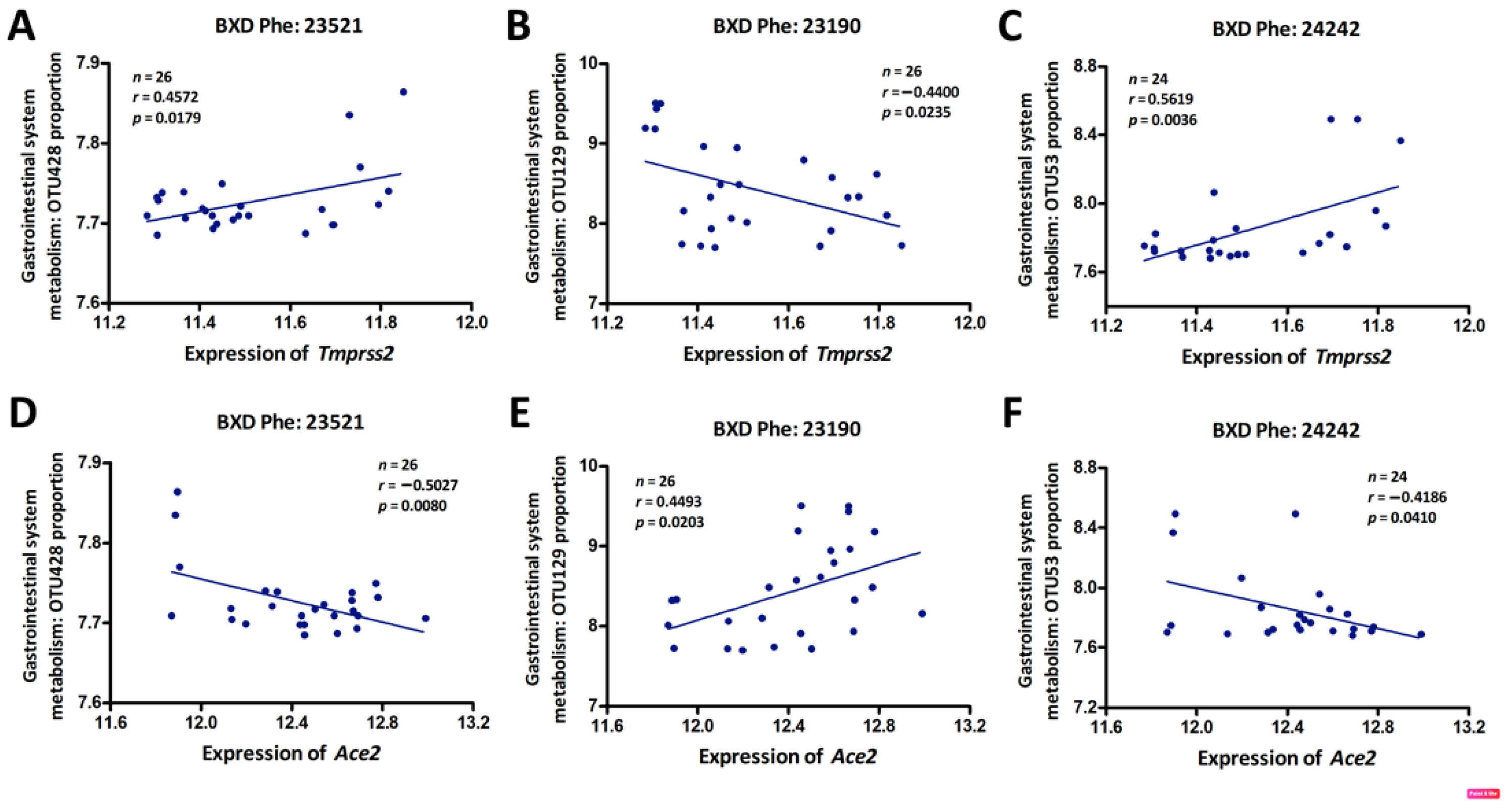
3.4. Genetic Correlations between Tmprss2 and Ace2 and GI Microbiota
3.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
3.6. Protein–Protein Interactions (PPI) Subnetwork
4. Discussion
4.1. Dhx32 Was the Upstream Regulator of Tmprss2 and Ace2
4.2. GI Microbiota and COVID-19
4.3. Circadian Rhythms Involved in GI Function and Contribution to COVID-19
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G.J.V. Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses 2020, 12, 372. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.H.; Lui, R.N.; Sung, J.J. Covid-19 and the digestive system. J. Gastroenterol. Hepatol. 2020, 35, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Fang, Y.-Y.; Deng, Y.; Liu, W.; Wang, M.-F.; Ma, J.-P.; Xiao, W.; Wang, Y.-N.; Zhong, M.-H.; Li, C.-H. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin. Med. J. 2020, 133, 1025. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Jiang, X.; Zhang, Z.; Huang, S.; Zhang, Z.; Fang, Z.; Gu, Z.; Gao, L.; Shi, H.; Mai, L. Gastrointestinal symptoms of 95 cases with SARS-CoV-2 infection. Gut 2020, 69, 997–1001. [Google Scholar] [CrossRef]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Samanta, J.; Dhar, J.; Khaliq, A.; Kochhar, R. 2019 Novel Coronavirus Infection: Gastrointestinal Manifestations. J. Dig. Endosc. 2020, 11, 13–18. [Google Scholar] [CrossRef]
- Ahmed, M. Coronavirus Disease 2019: A Gastroenterologist’s Perspective in May 2020. Gastroenterol. Res. 2020, 13, 89. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [Green Version]
- Burgueño, J.F.; Reich, A.; Hazime, H.; Quintero, M.A.; Fernandez, I.; Fritsch, J.; Santander, A.M.; Brito, N.; Damas, O.M.; Deshpande, A. Expression of SARS-CoV-2 Entry Molecules ACE2 and TMPRSS2 in the Gut of Patients With IBD. Inflamm. Bowel Dis. 2020, 26, 797–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, S.; Dijkman, R.; Habjan, M.; Heurich, A.; Gierer, S.; Glowacka, I.; Welsch, K.; Winkler, M.; Schneider, H.; Hofmann-Winkler, H.; et al. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J. Virol. 2013, 87, 6150–6160. [Google Scholar] [CrossRef] [Green Version]
- Qing, E.; Hantak, M.P.; Galpalli, G.G.; Gallagher, T. Evaluating MERS-CoV Entry Pathways. In MERS Coronavirus; Humana: New York, NY, USA, 2020; pp. 9–20. [Google Scholar]
- Bertram, S.; Glowacka, I.; Blazejewska, P.; Soilleux, E.; Allen, P.; Danisch, S.; Steffen, I.; Choi, S.-Y.; Park, Y.; Schneider, H.; et al. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J. Virol. 2010, 84, 10016–10025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, S.; Greer, B.; Hooper, J.; Zijlstra, A.; Walker, B.; Quigley, J.; Hawthorne, S. The membrane-anchored serine protease, TMPRSS2, activates PAR-2 in prostate cancer cells. Biochem. J. 2005, 388, 967–972. [Google Scholar] [CrossRef] [Green Version]
- Garg, M.; Royce, S.G.; Tikellis, C.; Shallue, C.; Batu, D.; Velkoska, E.; Burrell, L.M.; Patel, S.K.; Beswick, L.; Jackson, A. Imbalance of the renin–angiotensin system may contribute to inflammation and fibrosis in IBD: A novel therapeutic target? Gut 2020, 69, 841–851. [Google Scholar] [CrossRef]
- Ning, L.; Shan, G.; Sun, Z.; Zhang, F.; Xu, C.; Lou, X.; Li, S.; Du, H.; Chen, H.; Xu, G. Quantitative proteomic analysis reveals the deregulation of nicotinamide adenine dinucleotide metabolism and CD38 in inflammatory bowel disease. BioMed Res. Int. 2019, 2019, 3950628. [Google Scholar] [CrossRef] [Green Version]
- Ashbrook, D.G.; Arends, D.; Prins, P.; Mulligan, M.K.; Roy, S.; Williams, E.G.; Lutz, C.M.; Valenzuela, A.; Bohl, C.J.; Ingels, J.F. A platform for experimental precision medicine: The extended BXD mouse family. Cell Syst. 2021, 12, 235–247.e239. [Google Scholar] [CrossRef]
- Ashbrook, D.G.; Arends, D.; Prins, P.; Mulligan, M.K.; Roy, S.; Williams, E.G.; Lutz, C.M.; Valenzuela, A.; Bohl, C.J.; Ingels, J.F. The expanded BXD family of mice: A cohort for experimental systems genetics and precision medicine. bioRxiv 2019, 672097. [Google Scholar] [CrossRef]
- Peirce, J.L.; Lu, L.; Gu, J.; Silver, L.M.; Williams, R.W. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet. 2004, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Gao, J.; Munkhsaikhan, U.; Li, N.; Gu, Q.; Pierre, J.F.; Starlard-Davenport, A.; Towbin, J.A.; Cui, Y.; Purevjav, E.; et al. The genetic dissection of Ace2 expression variation in the heart of murine genetic reference population. Front. Cardiovasc. Med. 2020, 7, 186. [Google Scholar] [CrossRef] [PubMed]
- Chesler, E.J.; Lu, L.; Wang, J.; Williams, R.W.; Manly, K.F. WebQTL: Rapid exploratory analysis of gene expression and genetic networks for brain and behavior. Nat. Neurosci. 2004, 7, 485–486. [Google Scholar] [CrossRef]
- Schughart, K.; Williams, R.W. Systems Genetics: Methods and Protocols; Humana: New York, NY, USA, 2017; Volume 24. [Google Scholar]
- Bolstad, B.M.; Irizarry, R.A.; Åstrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesler, E.J.; Lu, L.; Shou, S.; Qu, Y.; Gu, J.; Wang, J.; Hsu, H.C.; Mountz, J.D.; Baldwin, N.E.; Langston, M.A. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat. Genet. 2005, 37, 233–242. [Google Scholar] [CrossRef]
- Geisert, E.E.; Lu, L.; Freeman-Anderson, N.E.; Templeton, J.P.; Nassr, M.; Wang, X.; Gu, W.; Jiao, Y.; Williams, R.W. Gene expression in the mouse eye: An online resource for genetics using 103 strains of mice. Mol. Vis. 2009, 15, 1730. [Google Scholar] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.J.M.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Yue, F.; Cheng, Y.; Breschi, A.; Vierstra, J.; Wu, W.; Ryba, T.; Sandstrom, R.; Ma, Z.; Davis, C.; Pope, B.D.J.N. A comparative encyclopedia of DNA elements in the mouse genome. Nature 2014, 515, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulligan, M.K.; Mozhui, K.; Prins, P.; Williams, R.W. GeneNetwork: A toolbox for systems genetics. In Systems Genetics; Humana: New York, NY, USA, 2017; pp. 75–120. [Google Scholar]
- Williams, R.W.; Williams, E.G. Resources for systems genetics. In Systems Genetics; Humana: New York, NY, USA, 2017; pp. 3–29. [Google Scholar]
- Haley, C.S.; Knott, S.A. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 1992, 69, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [Green Version]
- McKnite, A.M.; Perez-Munoz, M.E.; Lu, L.; Williams, E.G.; Brewer, S.; Andreux, P.A.; Bastiaansen, J.W.; Wang, X.; Kachman, S.D.; Auwerx, J.; et al. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS ONE 2012, 7, e39191. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Benner, M.J.; Hancock, R.E. NetworkAnalyst-integrative approaches for protein–protein interaction network analysis and visual exploration. Nucleic Acids Res. 2014, 42, W167–W174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orchard, S.; Kerrien, S.; Abbani, S.; Aranda, B.; Bhate, J.; Bidwell, S.; Bridge, A.; Briganti, L.; Brinkman, F.S.; Cesareni, G.; et al. Protein interaction data curation: The International Molecular Exchange (IMEx) consortium. Nat. Methods 2012, 9, 345–350. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Zhang, X.; Xu, H. Don’t overlook digestive symptoms in patients with 2019 novel coronavirus disease (COVID-19). Clin. Gastroenterol. Hepatol. 2020, 18, 1636. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhong, S.; Li, F.; Liu, G.; Wang, X.; Liu, Z. Clinical characteristics and risk factors of gastrointestinal symptoms in patients with novel coronavirus pneumonia in Xinyang, Henan province. Chin. J. Dig. 2020, 40, 219794934. [Google Scholar]
- Villapol, S. Gastrointestinal symptoms associated with COVID-19: Impact on the gut microbiome. Transl. Res. 2020, 226, 57–69. [Google Scholar] [CrossRef]
- Sun, J.-K.; Zou, L.; Liu, Y.; Zhang, W.-H.; Ki, J.-J.; Kan, X.-H.; Chen, J.-D.; Shi, Q.-K.; Yuan, S.-T.; Gu, W.; et al. Acute gastrointestinal injury in critically ill patients with coronavirus disease 2019 in Wuhan, China. World J. Gastroenterol. 2020, 26, 6087–6097. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Zhang, J.; Ma, X.; Tan, J.; Chen, L.; Liu, S.; Xin, Y.; Zhuang, L. ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID-19. Biomed. Pharmacother. 2020, 131, 110678. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darvish-Damavandi, M.; Laycock, J.; Ward, C.; van Driel, M.S.; Goldgraben, M.A.; Buczacki, S.J. An analysis of SARS-CoV-2 cell entry genes identifies the intestine and colorectal cancer as susceptible tissues. Br. J. Surg. 2020, 107, e452–e454. [Google Scholar] [CrossRef]
- Antikchi, M.H.; Neamatzadeh, H.; Ghelmani, Y.; Jafari-Nedooshan, J.; Dastgheib, S.A.; Kargar, S.; Noorishadkam, M.; Bahrami, R.; Jarahzadeh, M.H. The Risk and Prevalence of COVID-19 Infection in Colorectal Cancer Patients: A Systematic Review and Meta-analysis. J. Gastrointest. Cancer 2020, 52, 73–79. [Google Scholar] [CrossRef]
- Lin, H.; Liu, W.; Fang, Z.; Liang, X.; Li, J.; Bai, Y.; Lin, L.; You, H.; Pei, Y.; Wang, F. Overexpression of DHX32 contributes to the growth and metastasis of colorectal cancer. Sci. Rep. 2015, 5, 9247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Liang, X.; Huang, R.; Zhang, Z. Up-regulation and clinical relevance of novel helicase homologue DHX32 in colorectal cancer. J. Exp. Clin. Cancer Res. 2009, 28, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Fang, Z.; Su, Y.; Li, P.; Wang, J.; Liao, H.; Hu, Q.; Ye, C.; Fang, Y.; Luo, Q. DHX32 promotes angiogenesis in colorectal cancer through augmenting β-catenin signaling to induce expression of VEGFA. EBioMedicine 2017, 18, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aktaş, B.; Aslim, B. Gut-lung axis and dysbiosis in COVID-19. Turk. J. Biol. 2020, 44, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Bao, R.; Hernandez, K.; Huang, L.; Luke, J.J. ACE2 and TMPRSS2 expression by clinical, HLA, immune, and microbial correlates across 34 human cancers and matched normal tissues: Implications for SARS-COV-2 COVID-19. J. Immunother. Cancer 2020, 8, e001020. [Google Scholar] [CrossRef]
- Wang, B.; Huang, Y. Which type of cancer patients are more susceptible to the SARS-COX-2: Evidence from a meta-analysis and bioinformatics analysis. Crit. Rev. Oncol. Hematol. 2020, 153, 103032. [Google Scholar] [CrossRef]
- Perez-Munoz, M.E.; McKnite, A.M.; Williams, E.G.; Auwerx, J.; Williams, R.W.; Peterson, D.A.; Ciobanu, D.C. Diet modulates cecum bacterial diversity and physiological phenotypes across the BXD mouse genetic reference population. PLoS ONE 2019, 14, e0224100. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Penninger, J.M. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp. Physiol. 2008, 93, 543–548. [Google Scholar] [CrossRef]
- Herichova, I.; Zsoldosova, K.; Vesela, A.; Zeman, M. Effect of angiotensin II infusion on rhythmic clock gene expression and local renin-angiotensin system in the aorta of Wistar rats. Endocr. Regul. 2014, 48, 144–151. [Google Scholar] [CrossRef]
- Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. Circadian rhythms: A regulator of gastrointestinal health and dysfunction. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 411–424. [Google Scholar] [CrossRef]
- Diallo, A.B.; Gay, L.; Coiffard, B.; Leone, M.; Mezouar, S.; Mege, J.-L. Daytime variation in SARS-CoV-2 infection and cytokine production. Microb. Pathog. 2020, 158, 105067. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Brooks, T.G.; Grant, G.R.; FitzGerald, G.A. Accounting for Time: Circadian Rhythms in the Time of COVID-19. J. Biol. Rhythm. 2020, 36, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Bakhtazad, A.; Jafari, R.; Khaksari, M.; Khastar, H.; Salehi, M.; Jafarisani, M.; Garmabi, B. Paying Attention to Circadian Rhythms in the Treatment of COVID-19. Basic Clin. Neurosci. 2020, 11, 233–246. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Symbol | Location (Chr, Mb) | Mean Expression | Max LRS | Cis-eQTL | Tmprss2 -r | Ace2 -r | Nonsynonymous Variants |
|---|---|---|---|---|---|---|---|---|
| 18242 | Oat | Chr7: 132.558 | 12.3394 | 13.2 | × | 0.118 | −0.186 | × |
| 20231 | Nkx1–2 | Chr7: 132.596 | 8.0721 | 13.5 | × | −0.426 | 0.061 | × |
| 76429 | Lhpp | Chr7: 132.611 | 9.4769 | 12.9 | × | −0.35 | −0.156 | × |
| 77938 | Fam53b | Chr7: 132.712 | 9.5488 | 12.4 | × | 0.085 | 0.216 | × |
| 360216 | Zranb1 | Chr7: 132.950 | 8.2709 | 12 | × | 0.273 | 0.048 | × |
| 13017 | Ctbp2 | Chr7: 132.988 | 9.9881 | 12.2 | × | 0.693 | −0.412 | √ |
| 73808 | Tex36 | Chr7: 133.587 | 5.5396 | 8.9 | × | −0.105 | −0.049 | × |
| 214766 | Mmp21 | Chr7: 133.674 | 6.8655 | 11.7 | × | −0.319 | −0.009 | × |
| 22276 | Uros | Chr7: 133.686 | 8.2217 | 9.9 | × | 0.468 | −0.345 | × |
| 66165 | Bccip | Chr7: 133.709 | 8.0386 | 13.4 | × | 0.636 | −0.167 | × |
| 101437 | Dhx32 | Chr7: 133.721 | 9.7337 | 15 | √ | 0.681 | −0.582 | × |
| 66930 | Fank1 | Chr7: 133.777 | 7.2585 | 14.2 | × | −0.176 | −0.278 | × |
| 11489 | Adam12 | Chr7: 133.883 | 7.5060 | 16.9 | × | −0.246 | −0.144 | √ |
| 330662 | Dock1 | Chr7: 134.671 | 9.5446 | 12 | × | −0.521 | 0.325 | × |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, F.; Gao, J.; Orgil, B.-O.; Bajpai, A.K.; Gu, Q.; Purevjav, E.; Davenport, A.S.; Li, K.; Towbin, J.A.; Black, D.D.; et al. Ace2 and Tmprss2 Expressions Are Regulated by Dhx32 and Influence the Gastrointestinal Symptoms Caused by SARS-CoV-2. J. Pers. Med. 2021, 11, 1212. https://doi.org/10.3390/jpm11111212
Xu F, Gao J, Orgil B-O, Bajpai AK, Gu Q, Purevjav E, Davenport AS, Li K, Towbin JA, Black DD, et al. Ace2 and Tmprss2 Expressions Are Regulated by Dhx32 and Influence the Gastrointestinal Symptoms Caused by SARS-CoV-2. Journal of Personalized Medicine. 2021; 11(11):1212. https://doi.org/10.3390/jpm11111212
Chicago/Turabian StyleXu, Fuyi, Jun Gao, Buyan-Ochir Orgil, Akhilesh Kumar Bajpai, Qingqing Gu, Enkhsaikhan Purevjav, Athena S. Davenport, Kui Li, Jeffrey A. Towbin, Dennis D. Black, and et al. 2021. "Ace2 and Tmprss2 Expressions Are Regulated by Dhx32 and Influence the Gastrointestinal Symptoms Caused by SARS-CoV-2" Journal of Personalized Medicine 11, no. 11: 1212. https://doi.org/10.3390/jpm11111212