
SARS-CoV-2 Variants of Concern: Presumptive Identification via Sanger Sequencing Analysis of the Receptor Binding Domain (RBD) Region of the S Gene
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Specimens and Ethical Statement
2.2. In Silico Analysis
2.3. RNA Extraction and cDNA Synthesis
2.4. RBD Polymerase Chain Reaction (PCR) Amplification and Sanger Sequencing
2.5. Limit of Detection (LoD) and Repeatability
2.6. Bi-Directional Sanger Sequencing Analysis
3. Results
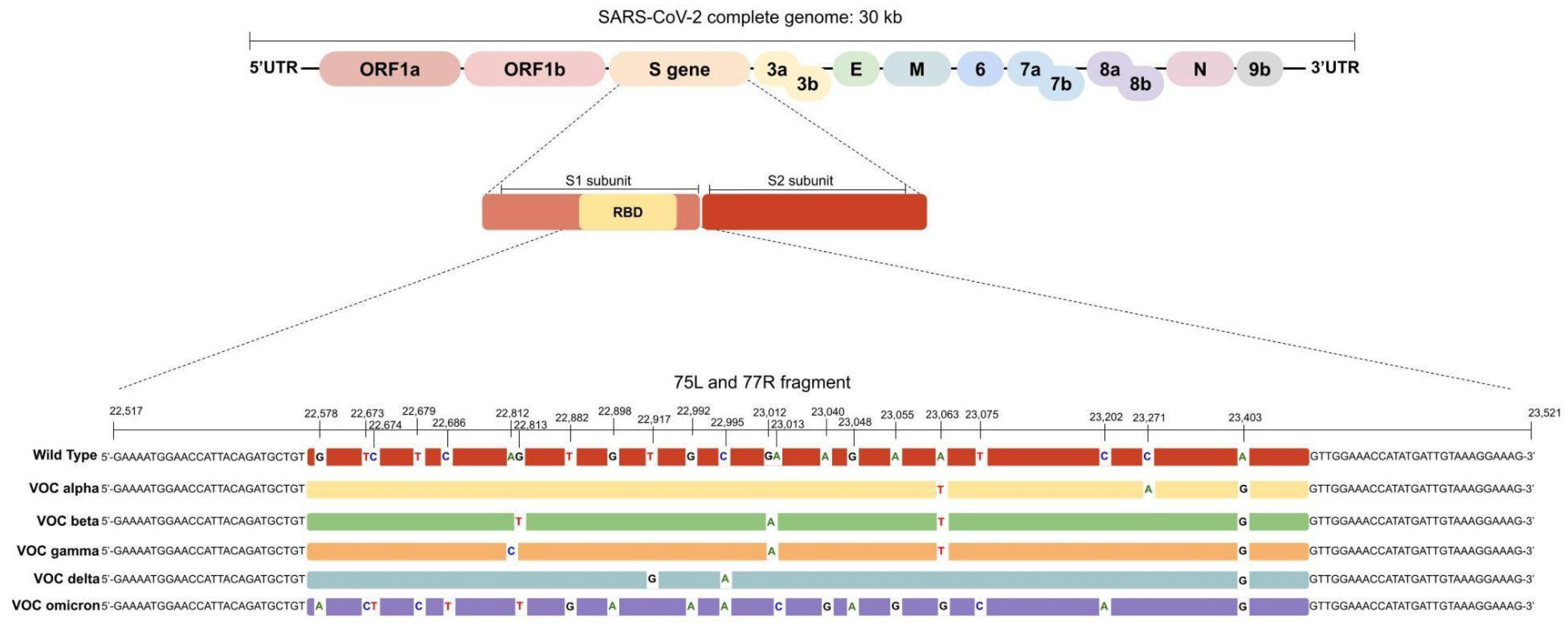
3.1. In Silico Analysis
3.2. Sanger Sequencing and WGS Results
3.3. Sanger Sequencing Quality
3.4. Limit of Detection
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cascella, M.; Rajnik, A.; Aleem, S.C.; Dulebohn, R. Di Napoli. Features, Evaluation, and Treatment of Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK554776/ (accessed on 12 August 2022).
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- WHO. Tracking SARS-CoV-2 Variants. 2022. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 6 June 2022).
- Song, S.; Ma, L.; Zou, D.; Tian, D.; Li, C.; Zhu, J.; Chen, M.; Wang, A.; Ma, Y.; Li, M.; et al. The Global Landscape of SARS-CoV-2 Genomes, Variants, and Haplotypes in 2019nCoVR. Genom. Proteom. Bioinform. 2020, 18, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.; Kronig, I.; Neher, R.A.; Eckerle, I.; Vetter, P.; Kaiser, L.; Geneva Center for Emerging Viral Diseases. Novel SARS-CoV-2 variants: The pandemics within the pandemic. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2021, 27, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef] [PubMed]
- Bechtold, P.; Wagner, P.; Hosch, S.; Siegrist, D.; Ruiz-Serrano, A.; Gregorini, M.; Mpina, M.; Ondó, F.A.; Obama, J.; Ayekaba, M.O.; et al. Rapid Identification of SARS-CoV-2 Variants of Concern Using a Portable peakPCR Platform. Anal. Chem. 2021, 93, 16350–16359. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next generation sequencing of SARS-CoV-2 genomes: Challenges, applications and opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Tétreault, M.; Dyment, D.A.; Zou, R.; Kernohan, K.; Geraghty, M.T.; FORGE Canada Consortium; Care4Rare Canada Consortium; Hartley, T.; Boycott, K.M. Concordance between whole-exome sequencing and clinical Sanger sequencing: Implications for patient care. Mol. Genet. Genom. Med. 2016, 4, 504–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorlass, E.G.; Lourenço, K.L.; Magalhães, R.; Sato, H.; Fiorini, A.; Peixoto, R.; Coelho, H.P.; Telezynski, B.L.; Scagion, G.P.; Ometto, T.; et al. Survey of SARS-CoV-2 genetic diversity in two major Brazilian cities using a fast and affordable Sanger sequencing strategy. Genomics 2021, 113, 4109–4115. [Google Scholar] [CrossRef] [PubMed]
- CDC. 2019-Novel Coronavirus (2019-nCoV) Real-Time RT-PCR Diagnostic Panel. Centers for Disease Control and Prevention. Available online: https://www.fda.gov/media/134922/download (accessed on 23 October 2022).
- ARTICnetwork Project. Available online: https://github.com/artic-network/artic-ncov2019/blob/7e359dae37d894b40ae7e35c3582f14244ef4d36/primer_schemes/nCoV-2019/V3/nCoV-2019.tsv (accessed on 6 June 2021).
- Wink, P.L.; Volpato, F.; Lima-Morales, D.; Paiva, R.M.; Willig, J.B.; Bock, H.; Paris, F.; Barth, A.L. RT-qPCR half-reaction optimization for the detection of SARS-CoV-2. Rev. Soc. Bras. Med. Trop. 2021, 54, e03192020. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H. A Routine Sanger Sequencing Target Specific Mutation Assay for SARS-CoV-2 Variants of Concern and Interest. Viruses 2021, 13, 2386. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, V.; Chrysovergis, A.; Ragos, V.; Tsiambas, E.; Katsinis, S.; Manoli, A.; Papouliakos, S.; Roukas, D.; Mastronikolis, S.; Peschos, D.; et al. From Delta to Omicron: S1-RBD/S2 mutation/deletion equilibrium in SARS-CoV-2 defined variants. Gene 2022, 814, 146134. [Google Scholar] [CrossRef] [PubMed]
- Crossley, B.M.; Bai, J.; Glaser, A.; Maes, R.; Porter, E.; Killian, M.L.; Clement, T.; Toohey-Kurth, K. Guidelines for Sanger sequencing and molecular assay monitoring. J. Vet. Diagn. Investig. 2020, 32, 767–775. [Google Scholar] [CrossRef] [PubMed]
- S Surface Glycoprotein of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/gene/43740568 (accessed on 6 October 2021).
- Kannan, S.; Shaik Syed Ali, P.; Sheeza, A. Omicron (B.1.1.529)-variant of concern-molecular profile and epidemiology: A mini review. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 8019–8022. [Google Scholar] [CrossRef] [PubMed]
- CoVariants. 2022. Available online: https://covariants.org/ (accessed on 10 December 2022).
- Boletim Genômico 14-Vigilância Genômica CEVS/SES-RS. Available online: https://coronavirus.rs.gov.br/upload/arquivos/202211/21171734-211122-informe-vigilancia-genomica.pdf (accessed on 10 December 2022).
- Daniels, R.S.; Harvey, R.; Ermetal, B.; Xiang, Z.; Galiano, M.; Adams, L.; McCauley, J.W. A Sanger sequencing protocol for SARS-CoV-2 S-gene. Influenza Other Respir. Viruses 2021, 15, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Aranha, C.; Patel, V.; Bhor, V.; Gogoi, D. Cycle threshold values in RT-PCR to determine dynamics of SARS-CoV-2 viral load: An approach to reduce the isolation period for COVID-19 patients. J. Med. Virol. 2021, 93, 6794–6797. [Google Scholar] [CrossRef] [PubMed]
- Fox-Lewis, A.; Fox-Lewis, S.; Beaumont, J.; Drinković, D.; Harrower, J.; Howe, K.; Jackson, C.; Rahnama, F.; Shilton, B.; Qiao, H.; et al. SARS-CoV-2 viral load dynamics and real-time RT-PCR cycle threshold interpretation in symptomatic non-hospitalised individuals in New Zealand: A multicentre cross sectional observational study. Pathology 2021, 53, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Gentile, A.; Juarez, M.; Lucion, M.F.; Pejito, M.N.; Alexay, S.; Orqueda, A.S.; Bollon, L.R.; Mistchenko, A. COVID-19 in Children: Correlation Between Epidemiologic, Clinical Characteristics, and RT-qPCR Cycle Threshold Values. Pediatr. Infect. Dis. J. 2022, 41, 666–670. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| WHO Variant of Concern | Genomic Annotation | Amino Acid Change | Pangolineage | Nextstrain Clade |
|---|---|---|---|---|
| Alpha | A23063T | N501Y | B.1.1.7 | 20I (V1) |
| C23271A | A570D | |||
| A23403G | D614G | |||
| Beta | G22813T | K417N | B.1.351 | 20H (V2) |
| G23012A | E484K | |||
| A23063T | N501Y | |||
| A23403G | D614G | |||
| Gamma | A22812C | K417T | P.1 | 20J (V3) |
| G23012A | E484K | |||
| A23063T | N501Y | |||
| A23403G | D614G | |||
| Delta | T22917G | L452R | B.1.617.2 | 21A, 21I, 21J |
| C22995A | T478K | |||
| A23403G | D614G | |||
| Omicron | G22578A | G339D | B.1.1.529 | 21K, 21L, 21M |
| T22673C | S371L | |||
| C22674T | ||||
| T22679C | S373P | |||
| C22686T | S375F | |||
| G22813T | K417N | |||
| T22882G | N440K | |||
| G22898A | G446S | |||
| G22992A | S477N | |||
| C22995A | T478K | |||
| A23013C | E484A | |||
| A23040G | Q493R | |||
| G23048A | G496S | |||
| A23055G | Q498R | |||
| A23063T | N501Y | |||
| T23075C | Y505H | |||
| C23202A | T547K | |||
| A23403G | D614G |
| BA.1 Omicron Descendent Lineage | BA.2 Omicron Descendent Lineage | BA.4 and 5 Omicron Descendent Lineage | BQ.1 Omicron Descendent Lineage | ||||
|---|---|---|---|---|---|---|---|
| Genome Annotation | Amino Acid Change | Genome Annotation | Amino Acid Change | Genome Annotation | Amino Acid Change | Genome Annotation | Amino Acid Change |
| G22578A | G339D | G22578A | G339D | G22578A | G339D | G22578A | G339D |
| - | R346T absent | - | R346T absent | - | R346T absent | G22599C | R346T |
| - | L368I absent | - | L368I absent | - | L368I absent | - | L368I absent |
| T22673C/ C22674T | S371L | C22674T | S371F | C22674T | S371F | C22674T | S371F |
| T22679C | S373P | T22679C | S373P | T22679C | S373P | T22679C | S373P |
| C22686T | S375F | C22686T | S375F | C22686T | S375F | C22686T | S375F |
| - | T376A absent | A22688G | T376A | A22688G | T376A | A22688G | T376A |
| - | D405N absent | G22775A | D405N | G22775A | D405N | G22775A | D405N |
| - | R408S absent | A22786C | R408S | A22786C | R408S | A22786C | R408S |
| G22813T | K417N | G22813T | K417N | G22813T | K417N | G22813T | K417N |
| T22882G | N440K | T22882G | N440K | T22882G | N440K | T22882G | N440K |
| - | K444T absent | - | K444T absent | - | K444T absent | A22893C | K444T |
| - | V445P absent | - | V445P absent | - | V445P absent | - | V445P absent |
| G22898A | G446S | - | G446S absent | - | G446S absent | - | G446S absent |
| - | L452R/Q absent | - | L452R/Q absent | T22917G | L452R | T22917G | L452R |
| - | N460K absent | - | N460K absent | - | N460K absent | T22942A | N460K |
| G22992A | S477N | G22992A | S477N | G22992A | S477N | G22992A | S477N |
| C22995A | T478K | C22995A | T478K | C22995A | T478K | C22995A | T478K |
| A23013C | E484A | A23013C | E484A | A23013C | E484A | A23013C | E484A |
| - | F486V/S absent | - | F486V/S absent | T23018G | F486V | T23018G | F486V |
| - | F490S absent | - | F490S absent | - | F490S absent | - | F490S absent |
| A23040G | Q493R | A23040G | Q493R | - | Q493R absent | - | Q493R absent |
| G23048A | G496S | - | G496S absent | - | G496S absent | - | G496S absent |
| A23055G | Q498R | A23055G | Q498R | A23055G | Q498R | A23055G | Q498R |
| A23063T | N501Y | A23063T | N501Y | A23063T | N501Y | A23063T | N501Y |
| T23075C | Y505H | T23075C | Y505H | T23075C | Y505H | T23075C | Y505H |
| C23202A | T547K | - | T547K absent | - | T547K absent | - | T547K absent |
| A23403G | D614G | A23403G | D614G | A23403G | D614G | A23403G | D614G |
| CDC RT-qPCR Ct | Fragment Size (bp) | Pairwise Sequence Alignment Score | SNVs Identified via Sanger Sequencing | Sanger Presumptive Identification | Lineage Assigned via WGS | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | N1 | N2 | FWD | REV | FWD | REV | PangoLineage | WHO VOCs | ||
| 1 | 14.99 | 15.16 | 922 | ND | 98% | ND | A22812C; G23012A; A23063T; A23403G | gamma | P.1 | gamma |
| 4 | 14.86 | 15.17 | 904 | 838 | 95% | 89% | A23403G | non-VOC | B.1.1.28 | non-VOC |
| 5 | 16.67 | 15.28 | 858 | 817 | 92% | 92% | A22812C; G23012A; A23063T; A23403G | gamma | P.1 | gamma |
| 6 | 15.04 | 15.48 | 898 | 868 | 97% | 93% | G23012A; A23403G | non-VOC/zeta | P.2 | non-VOC/zeta |
| 8 | 15.26 | 14.15 | 837 | 781 | 94% | 94% | G23012A; A23403G | non-VOC/zeta | P.2 | non-VOC/zeta |
| 11 | 16.48 | 16.56 | 898 | 848 | 95% | 91% | T22917A; T23031C; A23403G | non-VOC/lambda | C.37 | non-VOC/lambda |
| 12 | 13.18 | 13.85 | 910 | 865 | 96% | 95% | A22812C; G23012A; A23063T; A23403G | gamma | P.1 | gamma |
| 13 | 14.97 | 13.94 | 941 | 932 | 97% | 99% | T22917A; C22995A; A23403G | delta | B.1.617.2 | delta |
| 14 | 18.32 | 18.57 | 936 | 920 | 97% | 97% | T22917A; C22995A; A23403G | delta | B.617.2-like | delta |
| 15 | 12.03 | 12.41 | 933 | 921 | 98% | 98% | A23403G | não-VOC | B.1.1.29 | non-VOC |
| 22 | 14.44 | 15.24 | 929 | 934 | 97% | 96% | A22812C; G23012A; A23063T; A23403G | gamma | P.1 | gamma |
| 23 | 18.71 | 17.65 | 914 | 896 | 96% | 94% | A22812C; G23012A; A23063T; A23403G | gamma | P.1 | gamma |
| 30 | 13.29 | 12.2 | 928 | 920 | 98% | 95% | T22917A; C22995A; A23403G | delta | B.617.2-like | delta |
| 32 | 15.94 | 15.1 | 898 | 868 | 97% | 93% | G23012A; A23403G | non-VOC/zeta | P.2 | non-VOC/zeta |
| 33 | 12.11 | 12.03 | 887 | 890 | 95% | 95% | G23012A; A23403G | non-VOC/zeta | P.2 | non-VOC/zeta |
| 34 | 18.18 | 18.48 | 885 | 792 | 95% | 89% | A22812C; G23012A; A23063T; A23403G | gamma | P.1 | gamma |
| 35 | 13.65 | 13.41 | 886 | 850 | 93% | 93% | A23403G | non-VOC | B.1.1.28 | non-VOC |
| 39 | 20.16 | 19.14 | 897 | ND | 96% | ND | - | non-VOC | B.1.1.28 | non-VOC |
| 40 | 18.47 | 19.71 | 768 | 494 | 95% | 91% | A23403G | non-VOC | B.1.1.28 | non-VOC |
| 42 | 18.67 | 19.99 | 826 | 771 | 94% | 93% | A22812C; G23012A; A23063T; A23403G | gamma | P.1 | gamma |
| 47 | 13.48 | 14.15 | 893 | 898 | 96% | 94% | A22812C; G23012A; A23063T; A23403G | gamma | P.1 | gamma |
| 51 | 12 | 12.5 | 877 | 810 | 94% | 92% | G22578A; T22673C; C22674T; T22679C; C22686T; G22813T; T22882G; G22898A; G22992A; C22995A; A23013C; A23040G; G23048A; A23055G; A23063T; T23075C; C23202A; A23403G | omicron | B.1.1.529 | omicron |
| 52 | 13 | 14.3 | 877 | 867 | 92% | 93% | G22578A; T22673C; C22674T; T22679C; C22686T; G22813T; T22882G; G22898A; G22992A; C22995A; A23013C; A23040G; G23048A; A23055G; A23063T; T23075C; C23202A; A23403G | omicron | B.1.1.529 | omicron |
| 58 | 10.98 | 9.18 | 877 | 867 | 95% | 95% | G23012A; A23403G | non-VOC/zeta | P.2 | non-VOC/zeta |
| 59 | 19.15 | 19.32 | 705 | 837 | 92% | 94% | A23403G | non-VOC | B.1.1.28 | non-VOC |
| 60 | 18.08 | 19.32 | 886 | 889 | 94% | 94% | G22578A; T22673C; C22674T; T22679C; C22686T; G22813T; T22882G; G22898A; G22992A; C22995A; A23013C; A23040G; G23048A; A23055G; A23063T; T23075C; C23202A; A23403G | omicron | B.1.1.529 | omicron |
| 61 | 20.87 | 32.82 | 854 | 837 | 92% | 92% | G22578A; T22673C; C22674T; T22679C; C22686T; G22813T; T22882G; G22898A; G22992A; C22995A; A23013C; A23040G; G23048A; A23055G; A23063T; T23075C; C23202A; A23403G | omicron | B.1.1.529 | omicron |
| 62 | 23.79 | 29.02 | 879 | 369 | 95% | 93% | T22917A; C22995A; A23403G | delta | B.617.2-like | delta |
| 63 | 19.89 | 20.4 | ND | 692 | ND | 94% | T22917A; C22995A; A23403G | delta | B.617.2-like | delta |
| 64 | 19.86 | 30.81 | 880 | 831 | 93% | 92% | G22578A; T22673C; C22674T; T22679C; C22686T; G22813T; T22882G; G22898A; G22992A; C22995A; A23013C; A23040G; G23048A; A23055G; A23063T; T23075C; C23202A; A23403G | omicron | B.1.1.529 | omicron |
| 65 | 12.97 | 12.89 | 928 | 900 | 96% | 95% | T22917A; C22995A; A23403G | delta | B.617.2-like | delta |
| 66 | 17.38 | 22.78 | 839 | ND | 93% | ND | G22578A; T22673C; C22674T; T22679C; C22686T; G22813T; T22882G; G22898A; G22992A; C22995A; A23013C; A23040G; G23048A; A23055G; A23063T; T23075C; C23202A; A23403G | omicron | B.1.1.529 | omicron |
| 68 | 16.05 | 20.8 | 901 | 873 | 92% | 93% | G22578A; T22673C; C22674T; T22679C; C22686T; G22813T; T22882G; G22898A; G22992A; C22995A; A23013C; A23040G; G23048A; A23055G; A23063T; T23075C; C23202A; A23403G | omicron | B.1.1.529 | omicron |
| 69 | 19.14 | 29 | 879 | 890 | 95% | 93% | T22917A; C22995A; A23403G | delta | B.617.2-like | delta |
| 70 | ND | ND | 897 | ND | 93% | ND | G22599C, C22674T, T22679C, C22686T, A22688G, G22775A, A22786C, G22813T, T22882G, A22893C, T22917G, T22942A, G22992A, C22995A, A23013C, T23018G, A23055G, A23063T, T23075C, A23403G | omicron, sublinage BQ.1 | BQ.1 | omicron, sublinage BQ.1 |
| 73 | ND | ND | 876 | ND | 94% | ND | G22599C, C22674T, T22679C, C22686T, A22688G, G22775A, A22786C, G22813T, T22882G, A22893C, T22917G, T22942A, G22992A, C22995A, A23013C, T23018G, A23055G, A23063T, T23075C, A23403G | omicron, sublinage BQ.1 | BQ.1 | omicron, sublinage BQ.1 |
| 74 | ND | ND | 760 | ND | 91% | ND | G22599C, C22674T, T22679C, C22686T, A22688G, G22775A, A22786C, G22813T, T22882G, A22893C, T22917G, T22942A, G22992A, C22995A, A23013C, T23018G, A23055G, A23063T, T23075C | omicron, sublinage BQ.1 | BQ.1 | omicron, sublinage BQ.1 |
| Serial Dilution | Target | Ct Standard | VL Standard (Copies/uL) | Ct Sample | VLSample (Copies/uL) | RBD PCR Result |
|---|---|---|---|---|---|---|
| 1:1 | N1 | 23.41 | 100,000 | 13.28 | 60,740,000 | Positive |
| N2 | 23.56 | 100,000 | 15.04 | 15,915,000 | ||
| 1:10 | N1 | 26.86 | 10,000 | 17.25 | 4,709,500 | Positive |
| N2 | 26.87 | 10,000 | 18.48 | 1,918,500 | ||
| 1:100 | N1 | 30.1 | 1000 | 20.62 | 540,550 | Positive |
| N2 | 30.53 | 1000 | 22.4 | 171,750 | ||
| 1:1000 | N1 | 33.77 | 100 | 25.88 | 18,285 | Negative |
| N2 | 34.29 | 100 | 27.55 | 7252 | ||
| 1:10,000 | N1 | 38.18 | 10 | 30.9 | 742 | Negative |
| N2 | 38.57 | 10 | 33.11 | 245 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, G.M.; Volpato, F.C.Z.; Wink, P.L.; Paiva, R.M.; Barth, A.L.; de-Paris, F. SARS-CoV-2 Variants of Concern: Presumptive Identification via Sanger Sequencing Analysis of the Receptor Binding Domain (RBD) Region of the S Gene. Diagnostics 2023, 13, 1256. https://doi.org/10.3390/diagnostics13071256
Rodrigues GM, Volpato FCZ, Wink PL, Paiva RM, Barth AL, de-Paris F. SARS-CoV-2 Variants of Concern: Presumptive Identification via Sanger Sequencing Analysis of the Receptor Binding Domain (RBD) Region of the S Gene. Diagnostics. 2023; 13(7):1256. https://doi.org/10.3390/diagnostics13071256
Chicago/Turabian StyleRodrigues, Grazielle Motta, Fabiana Caroline Zempulski Volpato, Priscila Lamb Wink, Rodrigo Minuto Paiva, Afonso Luís Barth, and Fernanda de-Paris. 2023. "SARS-CoV-2 Variants of Concern: Presumptive Identification via Sanger Sequencing Analysis of the Receptor Binding Domain (RBD) Region of the S Gene" Diagnostics 13, no. 7: 1256. https://doi.org/10.3390/diagnostics13071256