
Construct ceRNA Network and Risk Model of Breast Cancer Using Machine Learning Methods under the Mechanism of Cuproptosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
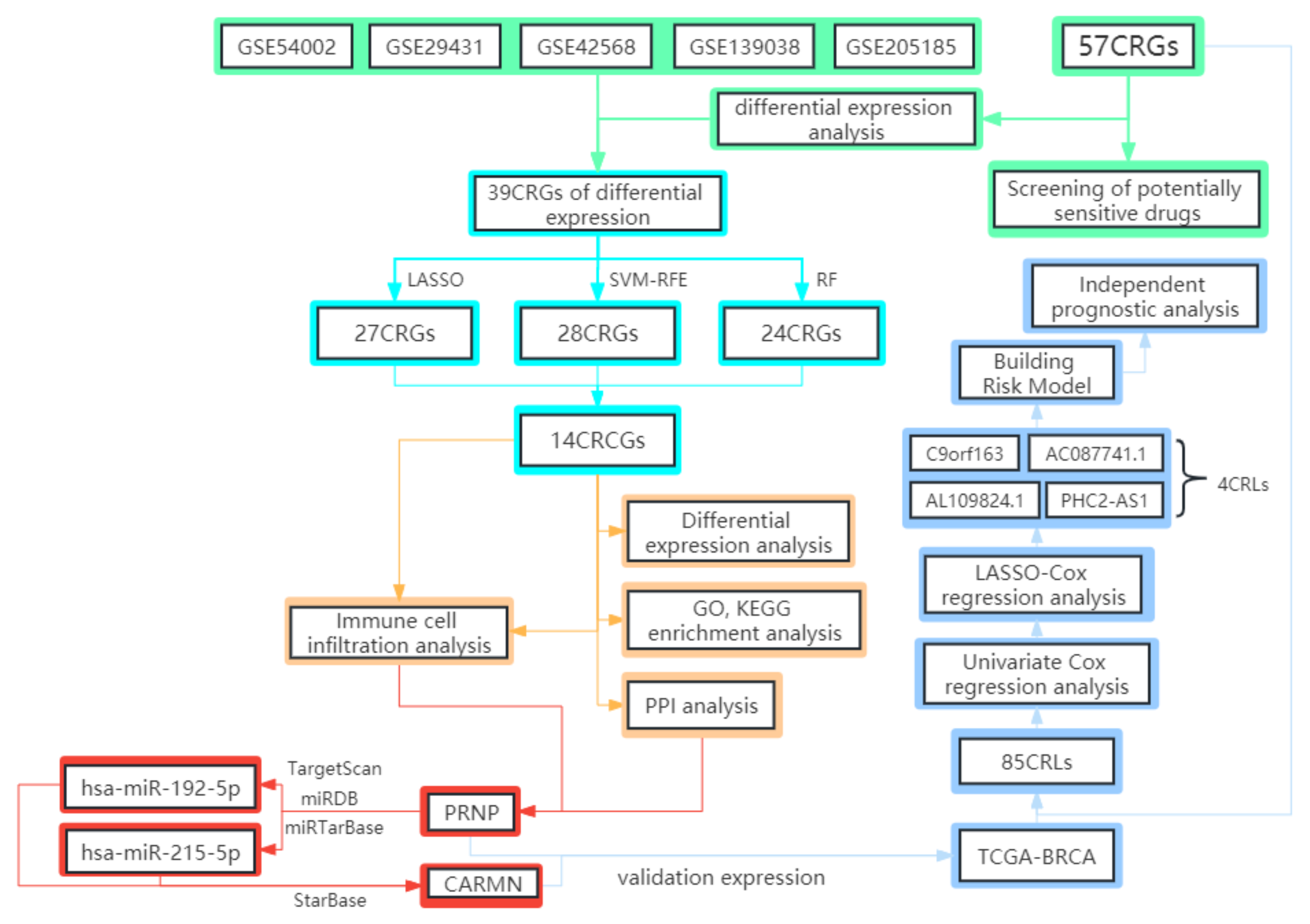
2. Materials and Methods
2.1. The Gathering and Pretreatment of Microarray Data
2.2. Obtainment, Differential Expression Analysis, and Correlation Analysis of CRGs
2.3. Screening of Cuproptosis-Related Characteristic Genes
2.4. GO, KEGG Enrichment Analysis
2.5. Immune Cell Infiltration Analysis and Correlation Analysis with CRCGs
2.6. Identification of Key CRCG and Construction of ceRNA Regulatory Network
2.7. Identification of CRLs and Construction of the Risk Model
2.8. Screening of Potentially Sensitive Medicines and Analyzing the Correlation between Drug Sensitivity and Risk Score
2.9. Statistical Analysis
3. Results
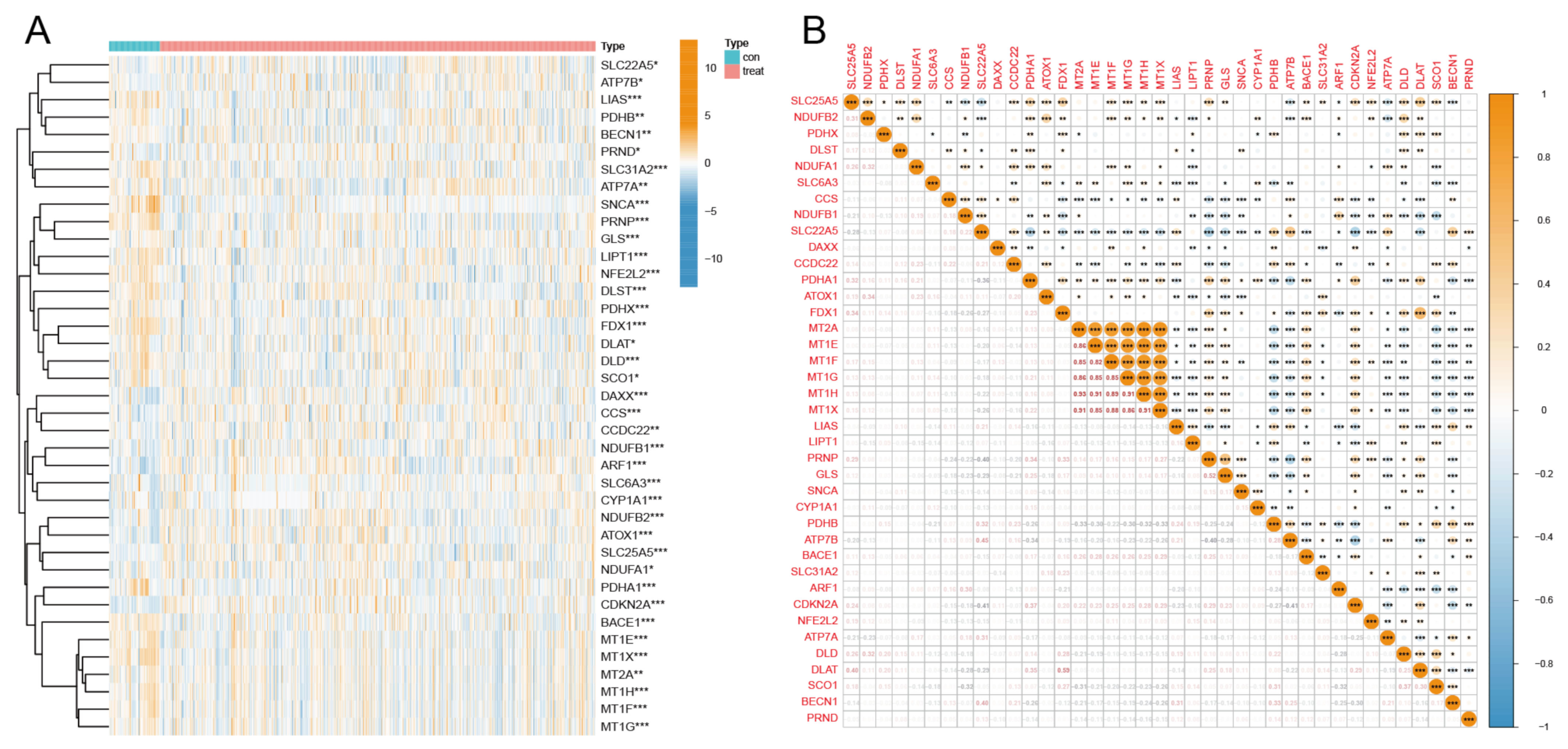
3.1. Differential Expression Analysis and Correlation Analysis of CRGs
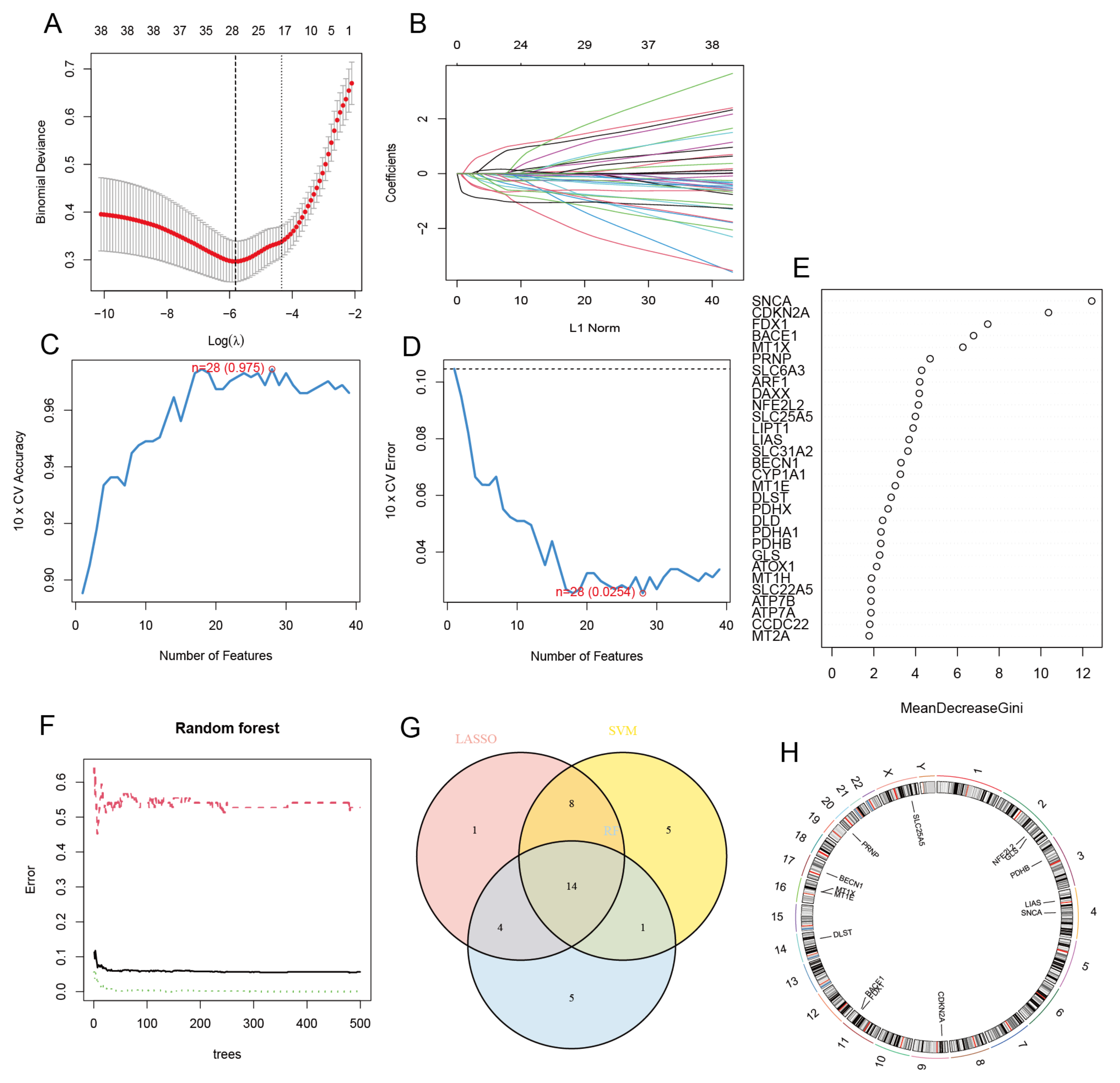
3.2. Selection of CRCGs via LASSO, SVM-RFE, and RF Methods
3.3. Differential Expression Analysis, GO, and KEGG Enrichment Analysis of CRCGs
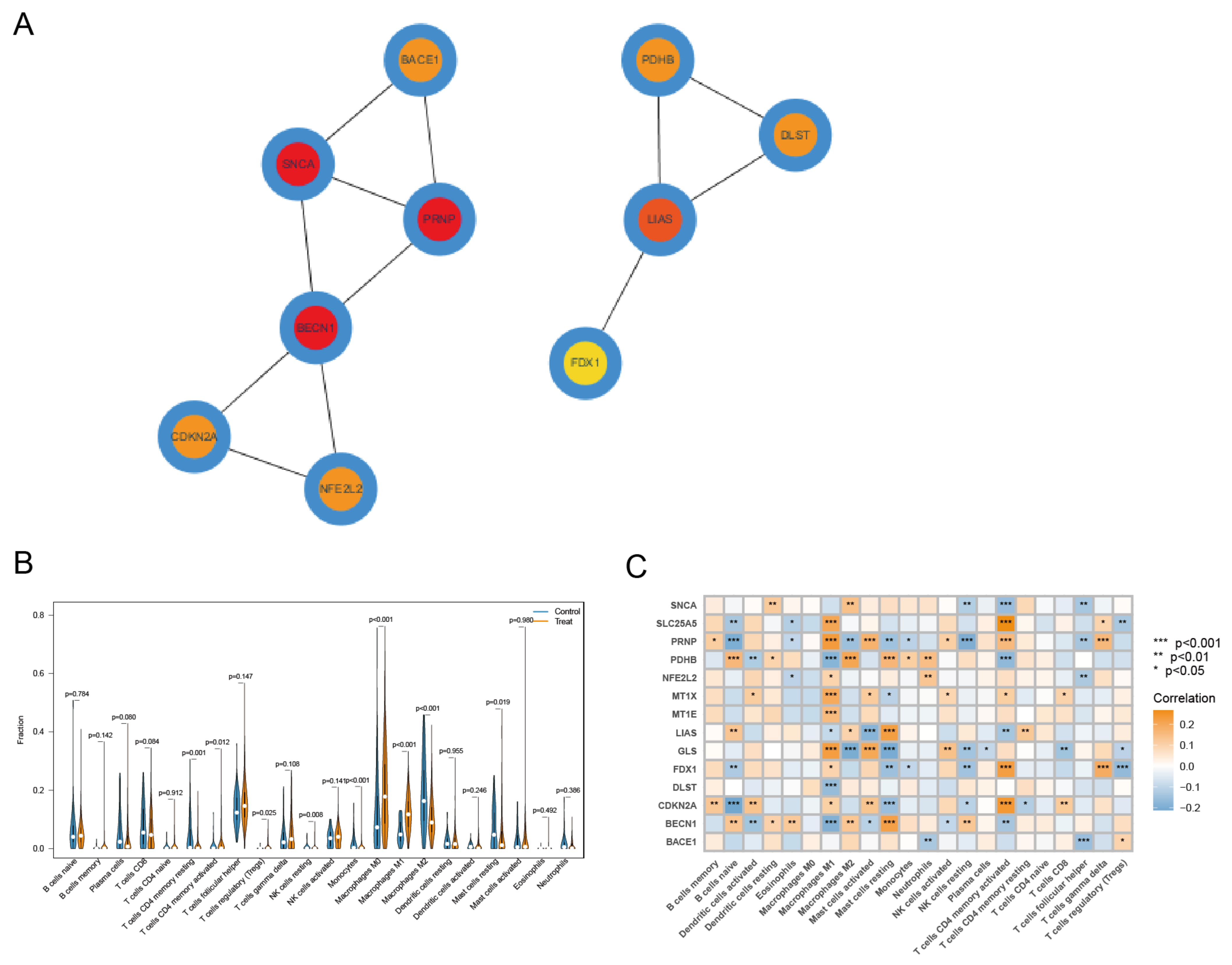
3.4. Identification of Key CRCG and Immune Cell Infiltration Analysis
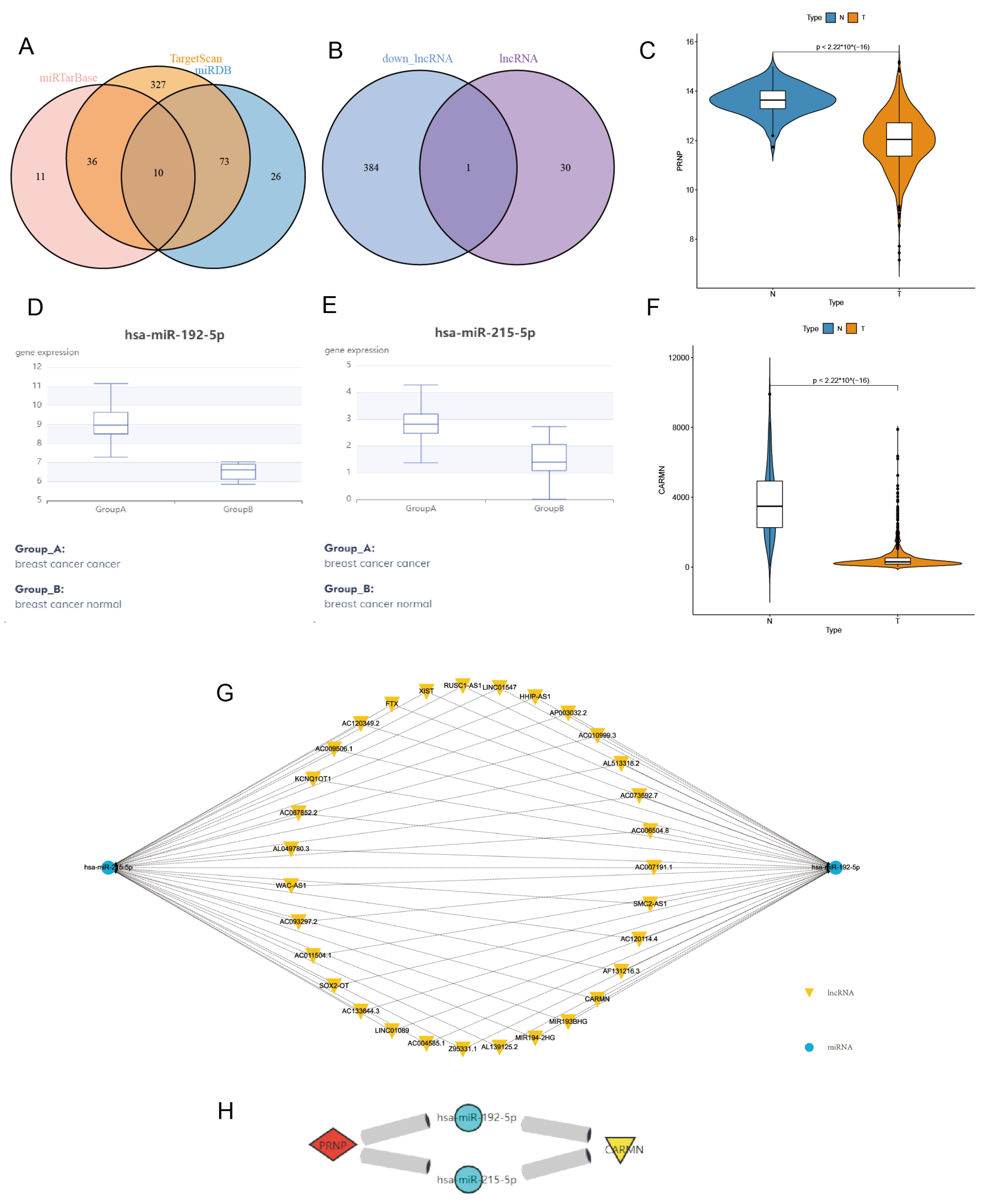
3.5. Construct and Verify PRNP-Related ceRNA Regulatory Network
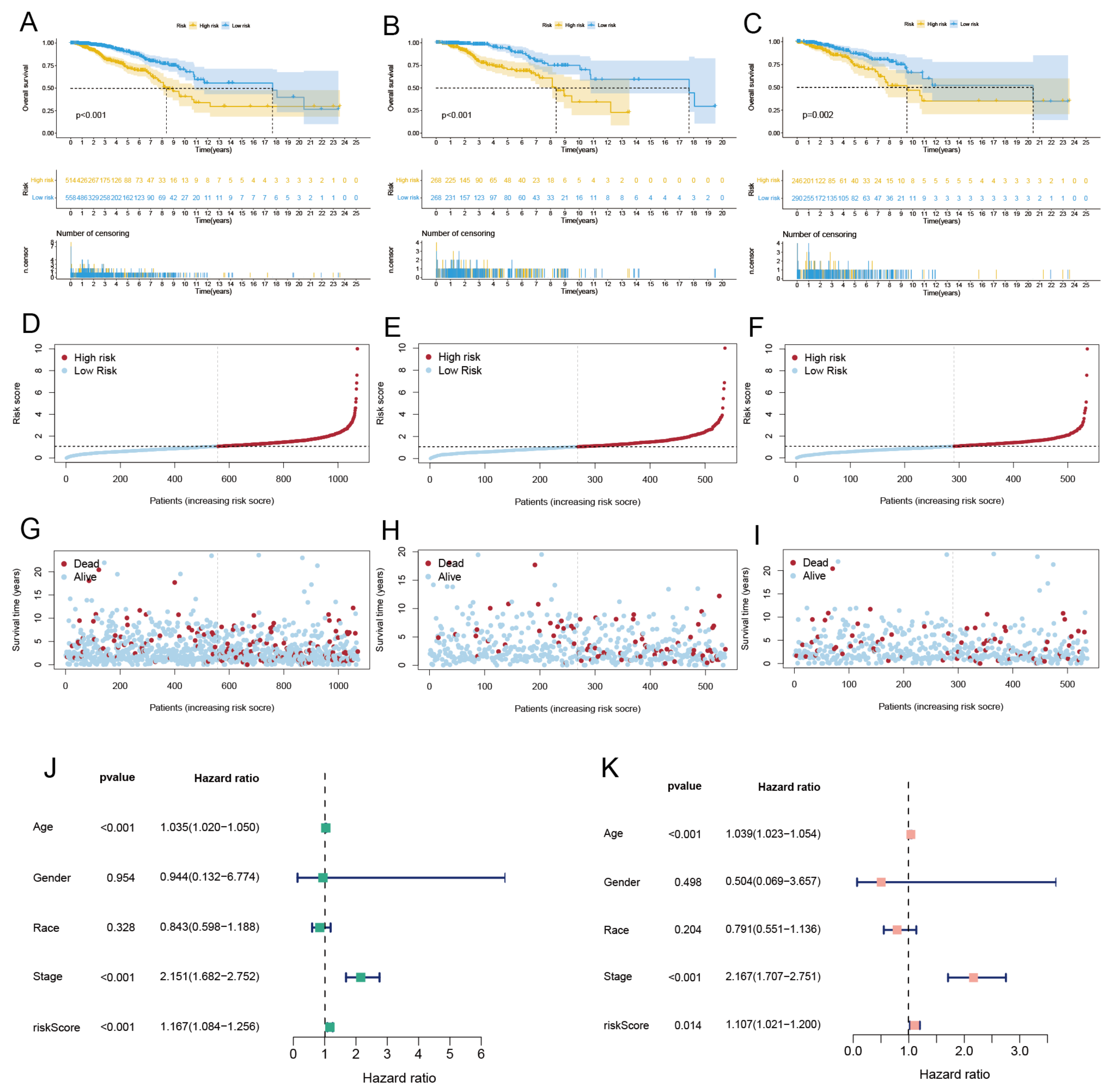
3.6. Identified CRLs and Constructed Risk Model
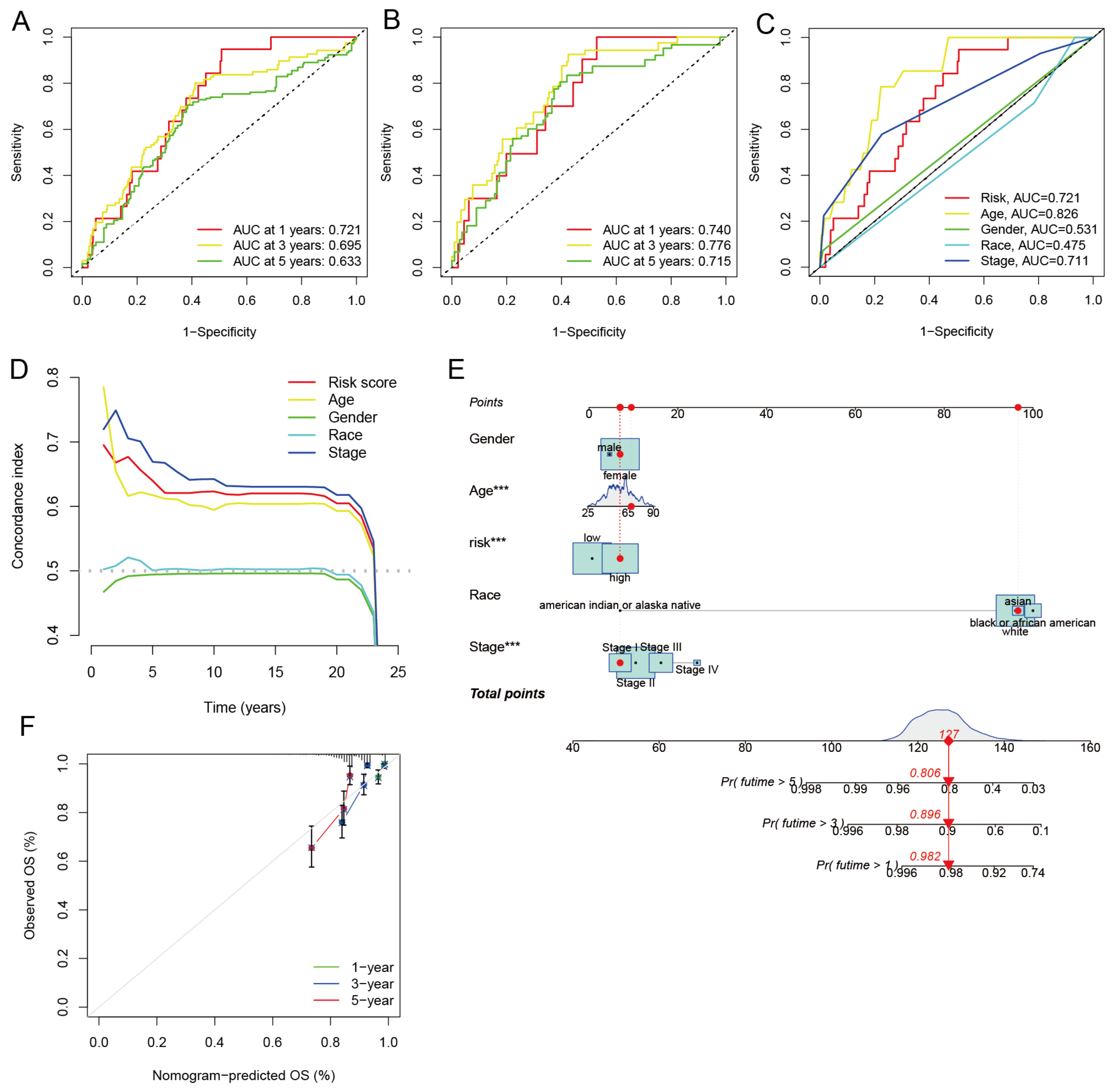
3.7. Independent Prognostic Analysis and Building a Nomogram
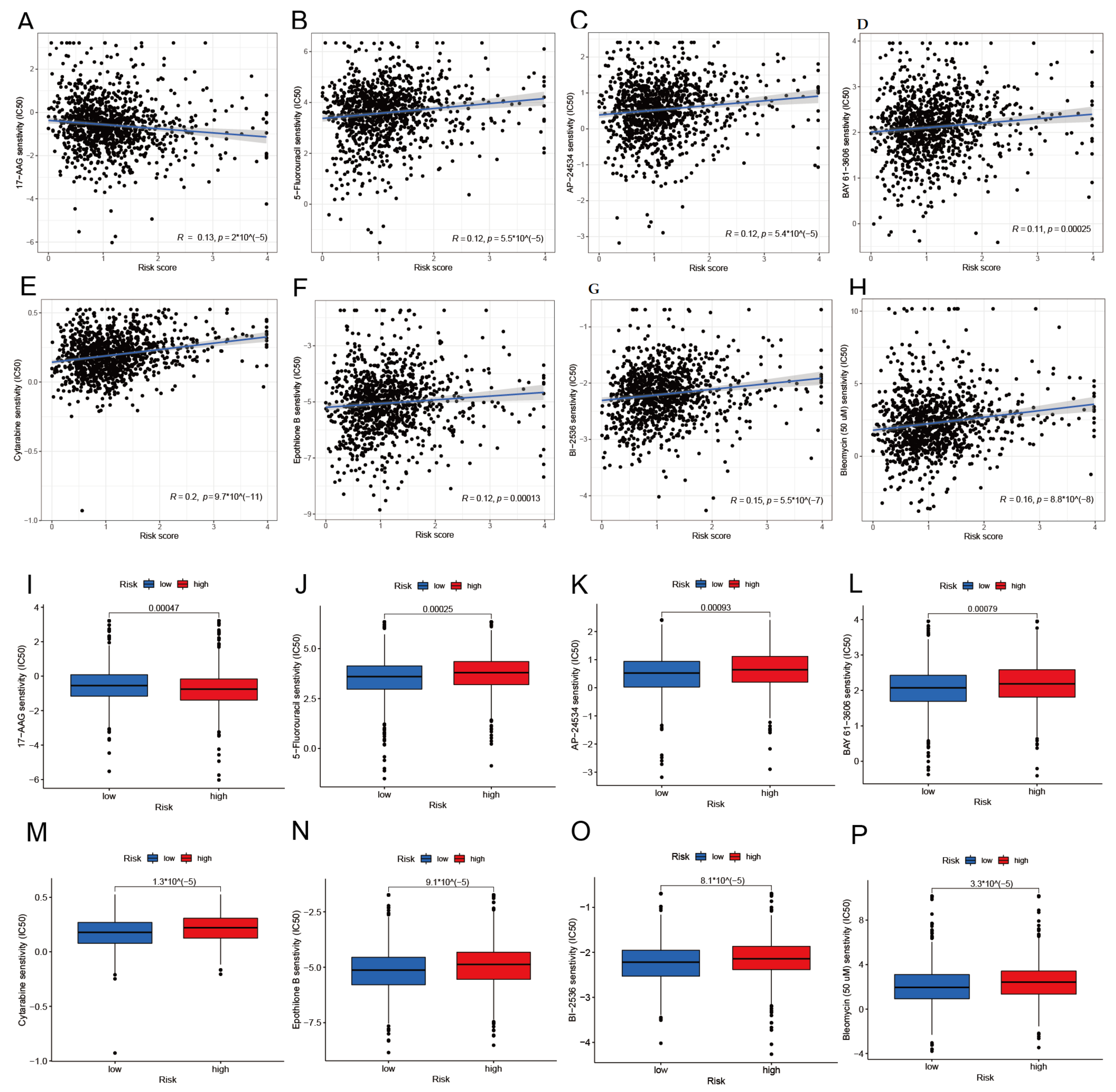
3.8. Drug Sensitivity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ceRNA | competing endogenous RNA |
| BRCA | breast cancer |
| GEO | gene expression omnibus |
| TCGA | The Cancer Genome Atlas |
| CRGs | cuproptosis-related genes |
| CRCGs | cuproptosis-related characteristic genes |
| LASSO | least absolute shrinkage and selection operator |
| SVM-RFE | recursive feature elimination by support vector machine |
| RF | random forest |
| PPI | protein–protein interaction |
| OS | overall survival |
| ROC | receiver operating characteristic curve |
| AUC | area under curve |
| TCA | tricarboxylic acid |
| MREs | miRNA response elements |
| CRLs | cuproptosis-related lncRNAs |
| FPKM | fragments per kilobase of transcript per million |
| DEGs | differential expression genes |
| LDA | linear discriminant analysis |
| MSE | mean squared error |
| C-index | concordance index |
| IC50 | the half maximal inhibitory concentration |
| HR | hazard ratios |
| CI | confidence intervals |
References
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, Y.; Shi, H.; Peng, Y.; Fan, X.; Li, C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pfl. Arch.-Eur. J. Physiol. 2020, 472, 1415–1429. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Vannini, I.; Fanini, F.; Fabbri, M. Emerging roles of microRNAs in cancer. Curr. Opin. Genet. Dev. 2018, 48, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.J.; Tay, Y. Noncoding RNA: RNA regulatory networks in cancer. Int. J. Mol. Sci. 2018, 19, 1310. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Zhu, H.; Feng, W.; Wan, Z.; Qi, X.; He, R.; Xie, L.; Li, Y. Database Mining Detected a Cuproptosis-Related Prognostic Signature and a Related Regulatory Axis in Breast Cancer. Dis. Markers 2022, 2022, 9004830. [Google Scholar] [CrossRef]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef]
- Park, M.K.; Zhang, L.; Min, K.W.; Cho, J.H.; Yeh, C.C.; Moon, H.; Hormaechea-Agulla, D.; Mun, H.; Ko, S.; Lee, J.W.; et al. NEAT1 is essential for metabolic changes that promote breast cancer growth and metastasis. Cell Metab. 2021, 33, 2380–2397. [Google Scholar] [CrossRef]
- Yue, X.; Wu, W.Y.; Dong, M.; Guo, M. LncRNA MALAT1 promotes breast cancer progression and doxorubicin resistance via regulating miR-570–3p. Biomed. J. 2021, 44, S296–S304. [Google Scholar] [CrossRef]
- Hu, J.; Huang, H.; Xi, Z.; Ma, S.; Ming, J.; Dong, F.; Guo, H.; Zhang, H.; Zhao, E.; Yao, G.; et al. LncRNA SEMA3B-AS1 inhibits breast cancer progression by targeting miR-3940/KLLN axis. Cell Death Dis. 2022, 13, 800. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.R.; Yang, L.H.; Jin, L.Z.; Yi, L.M.; Bing, P.P.; Zhou, J.; Yang, J.S. Identification of novel cuproptosis-related lncRNA signatures to predict the prognosis and immune microenvironment of breast cancer patients. Front. Oncol. 2022, 12, 988680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Y.; Bao, J.; Gao, W.; Wang, D.; Pan, H. Cuproptosis Combined with lncRNAs Predicts the Prognosis and Immune Microenvironment of Breast Cancer. Comput. Math. Methods Med. 2022, 2022, 5422698. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Huan, Y.; Li, Y.; Gao, X.; Sun, Q.; Zhang, F.; Jiang, T. Transcriptional and genetic alterations of cuproptosis-related genes correlated with malignancy and immune-infiltrate of esophageal carcinoma. Cell Death Discov. 2022, 8, 370. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhou, X.; Yang, L.; Zhou, H.; Meng, M.; Zhang, L.; Li, J. A Cuproptosis Activation Scoring model predicts neoplasm-immunity interactions and personalized treatments in glioma. Comput. Biol. Med. 2022, 148, 105924. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Dong, M.; Bi, Y.; Chen, S.; Huang, W.; Li, T.; Liu, B.; Fu, X.; Yang, Y. Identification and validation of a signature based on macrophage cell marker genes to predict recurrent miscarriage by integrated analysis of single-cell and bulk RNA-sequencing. Front. Immunol. 2022, 13, 1053819. [Google Scholar] [CrossRef] [PubMed]
- Sanz, H.; Valim, C.; Vegas, E.; Oller, J.M.; Reverter, F. SVM-RFE: Selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 2018, 19, 432. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Zhang, M.; Xie, P.; Wang, S.; Wang, Y. Comprehensive analysis of cuproptosis-related genes and tumor microenvironment infiltration characterization in breast cancer. Front. Immunol. 2022, 13, 978909. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, F. Identification of Ten-Gene Related to Lipid Metabolism for Predicting Overall Survival of Breast Invasive Carcinoma. Contrast Media Mol. Imaging 2022, 2022, 8348780. [Google Scholar] [CrossRef]
- Cheng, T.; Wu, Y.; Liu, Z.; Yu, Y.; Sun, S.; Guo, M.; Sun, B.; Huang, C. CDKN2A-mediated molecular subtypes characterize the hallmarks of tumor microenvironment and guide precision medicine in triple-negative breast cancer. Front. Immunol. 2022, 13, 970950. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.; Korm, S.; Karantanos, T.; Li, D.; Zhang, X.; Ritou, E.; Xu, H.; Lam, A.; English, J.; Zong, W.X.; et al. DLST-dependence dictates metabolic heterogeneity in TCA-cycle usage among triple-negative breast cancer. Commun. Biol. 2021, 4, 1289. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; He, Q.; Liu, W.; Liang, Q.; Peng, B.; Li, J.; Zhang, W.; Kang, F.; Hong, Q.; Yan, Y.; et al. Comprehensive analysis of the potential cuproptosis-related biomarker LIAS that regulates prognosis and immunotherapy of pan-cancers. Front. Oncol. 2022, 12, 952129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.x.; Zheng, H.; Tian, Y.; Luo, K.f.; Ma, S.j.; Wu, Z.w.; Tang, P.; Jiang, J.; Wang, M.H. SNCA inhibits epithelial-mesenchymal transition and correlates to favorable prognosis of breast cancer. Carcinogenesis 2022, 43, 1071–1082. [Google Scholar] [CrossRef]
- Ju, Q.; Li, X.; Zhang, H.; Yan, S.; Li, Y.; Zhao, Y. NFE2L2 is a potential prognostic biomarker and is correlated with immune infiltration in brain lower grade glioma: A pan-cancer analysis. Oxidative Med. Cell. Longev. 2020, 2020, 3580719. [Google Scholar] [CrossRef]
- Liu, H.; Ma, Y.; He, H.W.; Wang, J.P.; Jiang, J.D.; Shao, R.G. SLC9A3R1 stimulates autophagy via BECN1 stabilization in breast cancer cells. Autophagy 2015, 11, 2323–2334. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Yang, D.; Hou, Y.; Liu, S.; Zhao, M.; Qin, Y.; Chen, R.; Teng, Y.; Liu, M. Intracellular citrate accumulation by oxidized ATM-mediated metabolism reprogramming via PFKP and CS enhances hypoxic breast cancer cell invasion and metastasis. Cell Death Dis. 2019, 10, 228. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Sun, R.; Liu, L.; Yang, D.; Xiang, Q.; Li, L.; Tang, J.; Qiu, Z.; Peng, W.; Wang, Y.; et al. Tumor suppressor DRD2 facilitates M1 macrophages and restricts NF-κB signaling to trigger pyroptosis in breast cancer. Theranostics 2021, 11, 5214. [Google Scholar] [CrossRef]
- Rahal, O.M.; Wolfe, A.R.; Mandal, P.K.; Larson, R.; Tin, S.; Jimenez, C.; Zhang, D.; Horton, J.; Reuben, J.M.; McMurray, J.S.; et al. Blocking interleukin (IL) 4-and IL13-mediated phosphorylation of STAT6 (Tyr641) decreases M2 polarization of macrophages and protects against macrophage-mediated radioresistance of inflammatory breast cancer. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, S.; Wang, Q.; Zhang, X. Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J. Hematol. Oncol. 2017, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Liu, J.; Wang, X.; Yang, T.; Li, G.; Shang, Y.; Teng, X.; Yu, H.; Wang, S.; Huang, W. Identification of Key Transcription Factors and Immune Infiltration Patterns Associated With Breast Cancer Prognosis Using WGCNA and Cox Regression Analysis. Front. Oncol. 2021, 11, 5404. [Google Scholar] [CrossRef]
- Athanasios, A.; Charalampos, V.; Vasileios, T. Protein-protein interaction (PPI) network: Recent advances in drug discovery. Curr. Drug Metab. 2017, 18, 5–10. [Google Scholar] [CrossRef]
- Bernardino-Sgherri, J.; Siberchicot, C.; Auvré, F.; Busso, D.; Brocas, C.; El Masri, G.; Lioutsko, A.; Ferri, F.; Radicella, J.P.; Romeo, P.H.; et al. Tumor resistance to radiotherapy is triggered by an ATM/TAK1-dependent-increased expression of the cellular prion protein. Oncogene 2021, 40, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, S.; Ahmad, M.; Wu, S.; Zia, M.A.; Ahmed, I.; Iqbal, H.M.; Liu, Q.; Rehman, S.U. Cellular Prion Protein Role in Cancer Biology: Is It A Potential Therapeutic Target? Biomedicines 2022, 10, 2833. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.K.; Mathaiyan, J.; Kayal, S.; Nachiappa Ganesh, R. Identification of Differentially Expressed Mirna by Next Generation Sequencing in Locally Advanced Breast Cancer Patients of South Indian Origin. Asian Pac. J. Cancer Prev. 2022, 23, 2255–2261. [Google Scholar] [CrossRef]
- Sheng, X.; Dai, H.; Du, Y.; Peng, J.; Sha, R.; Yang, F.; Zhou, L.; Lin, Y.; Xu, S.; Wu, Y.; et al. LncRNA CARMN overexpression promotes prognosis and chemosensitivity of triple negative breast cancer via acting as miR143-3p host gene and inhibiting DNA replication. J. Exp. Clin. Cancer Res. 2021, 40, 205. [Google Scholar] [CrossRef] [PubMed]
- Déry, M.A.; Jodoin, J.; Ursini-Siegel, J.; Aleynikova, O.; Ferrario, C.; Hassan, S.; Basik, M.; LeBlanc, A.C. Endoplasmic reticulum stress induces PRNP prion protein gene expression in breast cancer. Breast Cancer Res. 2013, 15, R22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; Won, S.Y.; Jeong, B.H. Identification of prion disease-related somatic mutations in the prion protein gene (PRNP) in cancer patients. Cells 2020, 9, 1480. [Google Scholar] [CrossRef]
- Wiegmans, A.P.; Saunus, J.M.; Ham, S.; Lobb, R.; Kutasovic, J.R.; Dalley, A.J.; Miranda, M.; Atkinson, C.; Foliaki, S.T.; Ferguson, K.; et al. Secreted cellular prion protein binds doxorubicin and correlates with anthracycline resistance in breast cancer. JCI Insight 2019, 4, e124092. [Google Scholar] [CrossRef] [Green Version]
- Polley, M.Y.C.; Leon-Ferre, R.A.; Leung, S.; Cheng, A.; Gao, D.; Sinnwell, J.; Liu, H.; Hillman, D.W.; Eyman-Casey, A.; Gilbert, J.A.; et al. A clinical calculator to predict disease outcomes in women with triple-negative breast cancer. Breast Cancer Res. Treat. 2021, 185, 557–566. [Google Scholar] [CrossRef]
- Polley, M.Y.C.; Dickler, M.N.; Sinnwell, J.; Tenner, K.; de la Haba, J.; Loibl, S.; Goetz, M.P.; Bergh, J.; Roberston, J.; Couch, F.; et al. A clinical calculator to predict disease outcomes in women with hormone receptor-positive advanced breast cancer treated with first-line endocrine therapy. Breast Cancer Res. Treat. 2021, 189, 15–23. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Wu, B.; Du, J.; Li, L.; Zhao, J. Synergistic inhibition of the growth of MDA-MB-231 cells in triple-negative breast cancer by salinomycin combined with 17-AAG and its mechanism. Oncol. Lett. 2022, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Esgandari, K.; Mohammadian, M.; Zohdiaghdam, R.; Rastin, S.J.; Alidadi, S.; Behrouzkia, Z. Combined treatment with silver graphene quantum dot, radiation, and 17-AAG induces anticancer effects in breast cancer cells. J. Cell. Physiol. 2021, 236, 2817–2828. [Google Scholar] [CrossRef]
- Zheng, M.; Mei, Z.; Junaid, M.; Tania, M.; Fu, J.; Chen, H.C.; Khan, M.A. Synergistic Role of Thymoquinone on Anticancer Activity of 5-Fluorouracil in Triple Negative Breast Cancer Cells. Anti-Cancer Agents Med. Chem. Formerly Curr. Med. Chem. Anti-Cancer Agents 2022, 22, 1111–1118. [Google Scholar] [CrossRef]
- Kim, S.Y.; Park, S.E.; Shim, S.M.; Park, S.; Kim, K.K.; Jeong, S.Y.; Choi, E.K.; Hwang, J.J.; Jin, D.H.; Chung, C.D.; et al. Bay 61-3606 sensitizes TRAIL-induced apoptosis by downregulating Mcl-1 in breast cancer cells. PLoS ONE 2015, 10, e0146073. [Google Scholar] [CrossRef] [Green Version]
- Laakmann, E.; Witzel, I.; Müller, V. Efficacy of Liposomal Cytarabine in the treatment of leptomeningeal metastasis of breast cancer. Breast Care 2017, 12, 165–167. [Google Scholar] [CrossRef]
- Song, C.; Lowe, V.J.; Lee, S. Inhibition of Cdc20 suppresses the metastasis in triple negative breast cancer (TNBC). Breast Cancer 2021, 28, 1073–1086. [Google Scholar] [CrossRef]
- Jeong, S.B.; Im, J.H.; Yoon, J.H.; Bui, Q.T.; Lim, S.C.; Song, J.M.; Shim, Y.; Yun, J.; Hong, J.; Kang, K.W. Essential Role of Polo-like Kinase 1 (Plk1) Oncogene in Tumor Growth and Metastasis of Tamoxifen-Resistant Breast CancerRole of Plk1 in Tamoxifen-Resistant Breast Cancer. Mol. Cancer Ther. 2018, 17, 825–837. [Google Scholar] [CrossRef] [Green Version]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, J.; Fu, F.; Zhang, F.; Xia, Y.; Zhou, Y. Construct ceRNA Network and Risk Model of Breast Cancer Using Machine Learning Methods under the Mechanism of Cuproptosis. Diagnostics 2023, 13, 1203. https://doi.org/10.3390/diagnostics13061203
Deng J, Fu F, Zhang F, Xia Y, Zhou Y. Construct ceRNA Network and Risk Model of Breast Cancer Using Machine Learning Methods under the Mechanism of Cuproptosis. Diagnostics. 2023; 13(6):1203. https://doi.org/10.3390/diagnostics13061203
Chicago/Turabian StyleDeng, Jianzhi, Fei Fu, Fengming Zhang, Yuanyuan Xia, and Yuehan Zhou. 2023. "Construct ceRNA Network and Risk Model of Breast Cancer Using Machine Learning Methods under the Mechanism of Cuproptosis" Diagnostics 13, no. 6: 1203. https://doi.org/10.3390/diagnostics13061203