
The Role of Genetic Testing in Patients with Heritable Thoracic Aortic Diseases

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Methods
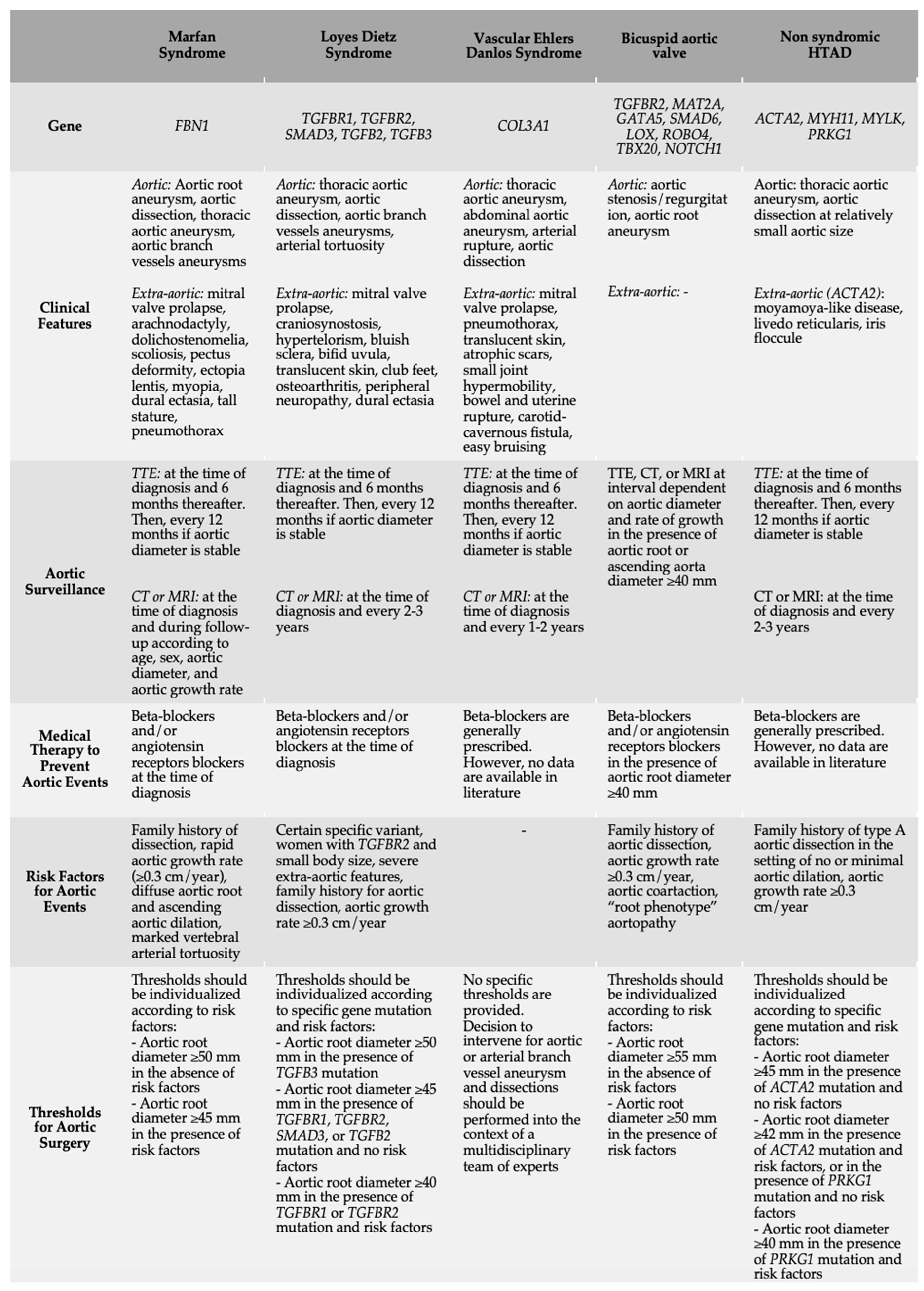
3. Syndromic HTAD
3.1. HTAD Related to Genes Encoding for Components of the Extracellular Matrix
3.1.1. Marfan Syndrome
3.1.2. Vascular Ehlers Danlos Syndrome
3.2. HTAD Related to Genes Encoding Component of the TGF-β
Loeys–Dietz Syndrome
4. Non-Syndromic HTAD
4.1. Bicuspid Aortic Valve
4.2. Other Non-Syndromic HTADs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Backer, J.; Bondue, A.; Budts, W.; Evangelista, A.; Gallego, P.; Jondeau, G.; Loeys, B.; Peña, M.L.; Teixido-Tura, G.; van de Laar, I.; et al. Genetic counselling and testing in adults with congenital heart disease: A consensus document of the ESC Working Group of Grown-Up Congenital Heart Disease, the ESC Working Group on Aorta and Peripheral Vascular Disease and the European Society of Human Genetics. Eur. J. Prev. Cardiol. 2020, 27, 1423–1435. [Google Scholar]
- Pyeritz, R.E. Heritable thoracic aortic disorders. Curr. Opin. Cardiol. 2014, 29, 97–102. [Google Scholar] [CrossRef]
- Albornoz, G.; Coady, M.A.; Roberts, M.; Davies, R.R.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Familial thoracic aortic aneurysms and dissections—Incidence, modes of inheritance, and phenotypic patterns. Ann. Thorac. Surg. 2006, 82, 1400–1405. [Google Scholar] [CrossRef]
- Baumgartner, H.; De Backer, J.; Babu-Narayan, S.V.; Budts, W.; Chessa, M.; Diller, G.P.; Lung, B.; Kluin, J.; Lang, I.M.; Meijboom, F.; et al. 2020 ESC Guidelines for the management of adult congenital heart disease. Eur. Heart. J. 2021, 42, 563–645. [Google Scholar] [CrossRef]
- Isselbacher, E.M.; Preventza, O.; Hamilton Black, J., 3rd; Augoustides, J.G.; Beck, A.W.; Bolen, M.A.; Braverman, A.C.; Bray, B.E.; Brown-Zimmerman, M.M.; Chen, E.P.; et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2022, 146, e334–e482. [Google Scholar] [CrossRef]
- Lopez-Sainz, A.; Mila, L.; Rodriguez-Palomares, J.; Limeres, J.; Granato, C.; La Mura, L.; Sabaté, A.; Guala, A.; Gutiérrez, L.; Galian-Gay, L.; et al. Aortic Branch Aneurysms and Vascular Risk in Patients with Marfan Syndrome. J. Am. Coll. Cardiol. 2021, 77, 3005–3012. [Google Scholar] [CrossRef]
- Regalado, E.S.; Morris, S.A.; Braverman, A.C.; Hostetler, E.M.; De Backer, J.; Li, R.; Pyeritz, R.E.; Yetman, A.T.; Cervi, E.; Shalhub, S.; et al. Comparative Risks of Initial Aortic Events Associated with Genetic Thoracic Aortic Disease. J. Am. Coll. Cardiol. 2022, 80, 857–869. [Google Scholar] [CrossRef]
- Baas, A.F.; Medic, J.; van’t Slot, R.; de Kovel, C.G.; Zhernakova, A.; Geelkerken, R.H.; Kranendonk, S.E.; van Sterkenburg, S.M.; Grobbee, D.E.; Boll, A.P.; et al. Association of the TGF-beta receptor genes with abdominal aortic aneurysm. Eur. J. Hum. Genet. 2010, 18, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Pannu, H.; Fadulu, V.T.; Chang, J.; Lafont, A.; Hasham, S.N.; Sparks, E.; Giampietro, P.F.; Zaleski, C.; Estrera, A.L.; Safi, H.J.; et al. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation 2005, 112, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Judge, D.P.; Dietz, H.C. Marfan’s syndrome. Lancet 2005, 366, 1965–1976. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Dietz, H.C.; Miller, D.C. Treatment of aortic disease in patients with Marfan syndrome. Circulation 2005, 111, e150–e157. [Google Scholar] [CrossRef]
- Monda, E.; Fusco, A.; Melis, D.; Caiazza, M.; Gragnano, F.; Mauriello, A.; Cirillo, A.; Rubino, M.; Esposito, A.; Grammegna, A.; et al. Clinical significance of family history and bicuspid aortic valve in children and young adult patients with Marfan syndrome. Cardiol. Young. 2020, 30, 663–667. [Google Scholar] [CrossRef]
- Fusco, A.; Mauriello, A.; Lioncino, M.; Palmiero, G.; Fratta, F.; Granato, C.; Cirillo, A.; Caiazza, M.; Monda, E.; Credendino, A.; et al. The Heart Muscle and Valve Involvement in Marfan Syndrome, Loeys-Dietz Syndromes, and Collagenopathies. Heart. Fail. Clin. 2022, 18, 165–175. [Google Scholar] [CrossRef]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shores, J.; Berger, K.R.; Murphy, E.A.; Pyeritz, R.E. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. N. Engl. J. Med. 1994, 330, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Teixido-Tura, G.; Forteza, A.; Rodríguez-Palomares, J.; González Mirelis, J.; Gutiérrez, L.; Sánchez, V.; Ibáñez, B.; García-Dorado, D.; Evangelista, A. Losartan Versus Atenolol for Prevention of Aortic Dilation in Patients With Marfan Syndrome. J. Am. Coll. Cardiol. 2018, 72, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Milleron, O.; Arnoult, F.; Ropers, J.; Aegerter, P.; Detaint, D.; Delorme, G.; Attias, D.; Tubach, F.; Dupuis-Girod, S.; Plauchu, H.; et al. Marfan Sartan: A randomized, double-blind, placebo-controlled trial. Eur. Heart. J. 2015, 36, 2160–2166. [Google Scholar] [CrossRef] [Green Version]
- Pitcher, A.; Spata, E.; Emberson, J.; Davies, K.; Halls, H.; Holland, L.; Wilson, K.; Reith, C.; Child, A.H.; Clayton, T.; et al. Angiotensin receptor blockers and β blockers in Marfan syndrome: An individual patient data meta-analysis of randomised trials. Lancet 2022, 400, 822–831. [Google Scholar] [CrossRef]
- Pelliccia, A.; Sharma, S.; Gati, S.; Bäck, M.; Börjesson, M.; Caselli, S.; Collet, J.P.; Corrado, D.; Drezner, J.A.; Halle, M.; et al. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease. Eur. Heart. J. 2021, 42, 17–96. [Google Scholar] [CrossRef]
- Monda, E.; Verrillo, F.; Rubino, M.; Palmiero, G.; Fusco, A.; Cirillo, A.; Caiazza, M.; Guarnaccia, N.; Mauriello, A.; Lioncino, M.; et al. Thoracic Aortic Dilation: Implications for Physical Activity and Sport Participation. Diagnostics 2022, 12, 1392. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.R.; Bristow, J. The Ehlers-Danlos syndrome: On beyond collagens. J. Clin. Investig. 2001, 107, 1063–1069. [Google Scholar] [CrossRef] [Green Version]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef]
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med. 2014, 16, 881–888. [Google Scholar] [CrossRef] [Green Version]
- Ong, K.T.; Perdu, J.; De Backer, J.; Bozec, E.; Collignon, P.; Emmerich, J.; Fauret, A.L.; Fiessinger, J.N.; Germain, D.P.; Georgesco, G.; et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: A prospective randomised, open, blinded-endpoints trial. Lancet 2010, 376, 1476–1484. [Google Scholar] [CrossRef]
- Frank, M.; Adham, S.; Seigle, S.; Legrand, A.; Mirault, T.; Henneton, P.; Albuisson, J.; Denarié, N.; Mazzella, J.M.; Mousseaux, E.; et al. Vascular Ehlers-Danlos Syndrome: Long-Term Observational Study. J. Am. Coll. Cardiol. 2019, 73, 1948–1957. [Google Scholar] [CrossRef]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef]
- Regalado, E.S.; Guo, D.C.; Villamizar, C.; Avidan, N.; Gilchrist, D.; McGillivray, B.; Clarke, L.; Bernier, F.; Santos-Cortez, R.L.; Leal, S.M.; et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ. Res. 2011, 109, 680–686. [Google Scholar] [CrossRef] [Green Version]
- Lynch, C.P.; Patel, M.; Seeley, A.H.; Seeley, M.A. Orthopaedic Management of Loeys-Dietz Syndrome: A Systematic Review. J. Am. Acad. Orthop. Surg. Glob. Res. Rev. 2021, 5, e21.00087. [Google Scholar] [CrossRef]
- Sousa, S.B.; Lambot-Juhan, K.; Rio, M.; Baujat, G.; Topouchian, V.; Hanna, N.; Le Merrer, M.; Brunelle, F.; Munnich, A.; Boileau, C.; et al. Expanding the skeletal phenotype of Loeys-Dietz syndrome. Am. J. Med. Genet. A 2011, 155A, 1178–1183. [Google Scholar] [CrossRef]
- Guerrerio, A.L.; Mateja, A.; Rasooly, M.; Levin, S.; Magnani, A.; Dempsey, C.; MacCarrick, G.; Dietz, H.C.; Brittain, E.; Boyce, A.M.; et al. Predictors of low bone density and fracture risk in Loeys-Dietz syndrome. Genet. Med. 2022, 24, 419–429. [Google Scholar] [CrossRef]
- MacCarrick, G.; Black, J.H., 3rd; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C., 3rd. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef]
- Mori, R.; Matsumoto, H.; Muro, S.; Morisaki, H.; Otsuki, R. Loeys-Dietz Syndrome Presenting with Giant Bullae and Asthma. J. Allergy Clin. Immunol. Pract. 2020, 8, 2058–2059. [Google Scholar] [CrossRef]
- Spinardi, L.; Vornetti, G.; De Martino, S.; Golfieri, R.; Faccioli, L.; Pastore Trossello, M.; Graziano, C.; Mariucci, E.; Donti, A. Intracranial Arterial Tortuosity in Marfan Syndrome and Loeys-Dietz Syndrome: Tortuosity Index Evaluation Is Useful in the Differential Diagnosis. Am. J. Neuroradiol. 2020, 41, 1916–1922. [Google Scholar] [CrossRef]
- Hughes, G.C. Aggressive aortic replacement for Loeys-Dietz syndrome. Tex. Heart. Inst. J. 2011, 38, 663–666. [Google Scholar]
- Gallo, E.M.; Loch, D.C.; Habashi, J.P.; Calderon, J.F.; Chen, Y.; Bedja, D.; van Erp, C.; Gerber, E.E.; Parker, S.J.; Sauls, K.; et al. Angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J. Clin. Investig. 2014, 124, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Sandor, G.G.; Alghamdi, M.H.; Raffin, L.A.; Potts, M.T.; Williams, L.D.; Potts, J.E.; Kiess, M.; van Breemen, C. A randomized, double blind pilot study to assess the effects of losartan vs. atenolol on the biophysical properties of the aorta in patients with Marfan and Loeys-Dietz syndromes. Int. J. Cardiol. 2015, 179, 470–475. [Google Scholar] [CrossRef]
- Zhou, D.; Feng, H.; Yang, Y.; Huang, T.; Qiu, P.; Zhang, C.; Olsen, T.R.; Zhang, J.; Chen, Y.E.; Mizrak, D.; et al. hiPSC Modeling of Lineage-Specific Smooth Muscle Cell Defects Caused by TGFBR1A230T Variant, and Its Therapeutic Implications for Loeys-Dietz Syndrome. Circulation 2021, 144, 1145–1159. [Google Scholar] [CrossRef]
- Iqbal, R.; Alom, S.; BinSaeid, J.; Harky, A. Loeys-Dietz syndrome pathology and aspects of cardiovascular management: A systematic review. Vascular 2021, 29, 3–14. [Google Scholar] [CrossRef]
- Jondeau, G.; Ropers, J.; Regalado, E.; Braverman, A.; Evangelista, A.; Teixedo, G.; De Backer, J.; Muiño-Mosquera, L.; Naudion, S.; Zordan, C.; et al. International Registry of Patients Carrying TGFBR1 or TGFBR2 Mutations: Results of the MAC (Montalcino Aortic Consortium). Circ. Cardiovasc. Genet. 2016, 9, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Aftab, M.; Cikach, F.S.; Zhu, Y.; Idrees, J.J.; Rigelsky, C.M.; Kalahasti, V.; Roselli, E.E.; Svensson, L.G. Loeys-Dietz syndrome: Intermediate-term outcomes of medically and surgically managed patients. J. Thorac. Cardiovasc. Surg. 2019, 157, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Michelena, H.I.; Della Corte, A.; Evangelista, A.; Maleszewski, J.J.; Edwards, W.D.; Roman, M.J.; Devereux, R.B.; Fernández, B.; Asch, F.M.; Barker, A.J.; et al. International Consensus Statement on Nomenclature and Classification of the Congenital Bicuspid Aortic Valve and Its Aortopathy, for Clinical, Surgical, Interventional and Research Purposes. Ann. Thorac. Surg. 2021, 112, e203–e235. [Google Scholar] [CrossRef]
- Yang, L.T.; Tribouilloy, C.; Masri, A.; Bax, J.J.; Delgado, V.; Girdauskas, E.; Evangelista, A.; Sundt, T.M., 3rd; Svensson, L.G.; Enriquez-Sarano, M.; et al. Clinical presentation and outcomes of adults with bicuspid aortic valves: 2020 update. Prog. Cardiovasc. Dis. 2020, 63, 434–441. [Google Scholar] [CrossRef]
- Galian-Gay, L.; Carro Hevia, A.; Teixido-Turà, G.; Rodríguez Palomares, J.; Gutiérrez-Moreno, L.; Maldonado, G.; Gonzàlez-Alujas, M.T.; Sao-Aviles, A.; Gallego, P.; Calvo-Iglesias, F.; et al. Familial clustering of bicuspid aortic valve and its relationship with aortic dilation in first-degree relatives. Heart 2019, 105, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Jaimes, K.; Prakash, S.K. Genetics in bicuspid aortic valve disease: Where are we? Prog. Cardiovasc. Dis. 2020, 63, 398–406. [Google Scholar] [CrossRef]
- Fatehi Hassanabad, A.; King, M.A.; Di Martino, E.; Fedak, P.W.M.; Garcia, J. Clinical implications of the biomechanics of bicuspid aortic valve and bicuspid aortopathy. Front. Cardiovasc. Med. 2022, 9, 922353. [Google Scholar] [CrossRef]
- Evangelista Masip, A.; Galian-Gay, L.; Guala, A.; Lopez-Sainz, A.; Teixido-Turà, G.; Ruiz Muñoz, A.; Valente, F.; Gutierrez, L.; Fernandez-Galera, R.; Casas, G.; et al. Unraveling Bicuspid Aortic Valve Enigmas by Multimodality Imaging: Clinical Implications. J. Clin. Med. 2022, 11, 456. [Google Scholar] [CrossRef]
- Zheng, Y.; Yang, K.; Chen, X.; Li, R.; Su, G.; Yin, G.; Wang, K.; Lu, M.; Zhao, S. Prognostic significance of myocardial fibrosis and CMR characteristics in bicuspid aortic valve with moderate and severe aortic insufficiency. Eur. Radiol. 2021, 31, 7262–7272. [Google Scholar] [CrossRef]
- Regitz-Zagrosek, V.; Roos-Hesselink, J.W.; Bauersachs, J.; Blomström-Lundqvist, C.; Cífková, R.; De Bonis, M.; Iung, B.; Johnson, M.R.; Kintscher, U.; Kranke, P.; et al. 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy. Eur. Heart. J. 2018, 39, 3165–3241. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Trybus, K.M.; Guo, D.C.; Sweeney, H.L.; Regalado, E.; Kamm, K.; Stull, J.T. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Regalado, E.S.; Guo, D.C.; Prakash, S.; Bensend, T.A.; Flynn, K.; Estrera, A.; Safi, H.; Liang, D.; Hyland, J.; Child, A.; et al. Aortic Disease Presentation and Outcome Associated With ACTA2 Mutations. Circ. Cardiovasc. Genet. 2015, 8, 457–464. [Google Scholar] [CrossRef] [Green Version]
- van de Laar, I.M.B.H.; Arbustini, E.; Loeys, B.; Björck, E.; Murphy, L.; Groenink, M.; Kempers, M.; Timmermans, J.; Roos-Hesselink, J.; Benke, K.; et al. European reference network for rare vascular diseases (VASCERN) consensus statement for the screening and management of patients with pathogenic ACTA2 variants. Orphanet J. Rare Dis. 2019, 14, 264. [Google Scholar] [CrossRef]


{kind=link}
| BAV Type | Presentation | Valve Disfunction | Aortopathy | Main Complications | Prognosis |
|---|---|---|---|---|---|
| Complex | Genetic syndromes and congenital heart lesions | Early and rapid | Early and rapid | Aortic dissection Endocarditis | Life expectancy may be reduced |
| Typical | Isolated BAV | Progressive | Progressive | Aortic dissection Endocarditis | Life expectancy usually preserved |
| Uncomplicated/ Undiagnosed | Isolated BAV | Mild and/or not progressive | Mild and/or not progressive | Typically silent condition | Excellent |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monda, E.; Lioncino, M.; Verrillo, F.; Rubino, M.; Caiazza, M.; Mauriello, A.; Guarnaccia, N.; Fusco, A.; Cirillo, A.; Covino, S.; et al. The Role of Genetic Testing in Patients with Heritable Thoracic Aortic Diseases. Diagnostics 2023, 13, 772. https://doi.org/10.3390/diagnostics13040772
Monda E, Lioncino M, Verrillo F, Rubino M, Caiazza M, Mauriello A, Guarnaccia N, Fusco A, Cirillo A, Covino S, et al. The Role of Genetic Testing in Patients with Heritable Thoracic Aortic Diseases. Diagnostics. 2023; 13(4):772. https://doi.org/10.3390/diagnostics13040772
Chicago/Turabian StyleMonda, Emanuele, Michele Lioncino, Federica Verrillo, Marta Rubino, Martina Caiazza, Alfredo Mauriello, Natale Guarnaccia, Adelaide Fusco, Annapaola Cirillo, Simona Covino, and et al. 2023. "The Role of Genetic Testing in Patients with Heritable Thoracic Aortic Diseases" Diagnostics 13, no. 4: 772. https://doi.org/10.3390/diagnostics13040772