
Cell-Free DNA Fragmentation Patterns in a Cancer Cell Line


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culturing and Processing of Supernatant
2.2. Cell-Free DNA Isolation
2.3. Cell-Free DNA Quantification
2.4. Cell-Free DNA Size Analysis
2.4.1. Automated Size Analysis of cfDNA
2.4.2. Agarose Gel Electrophoresis
2.5. Statistics
3. Results and Discussion
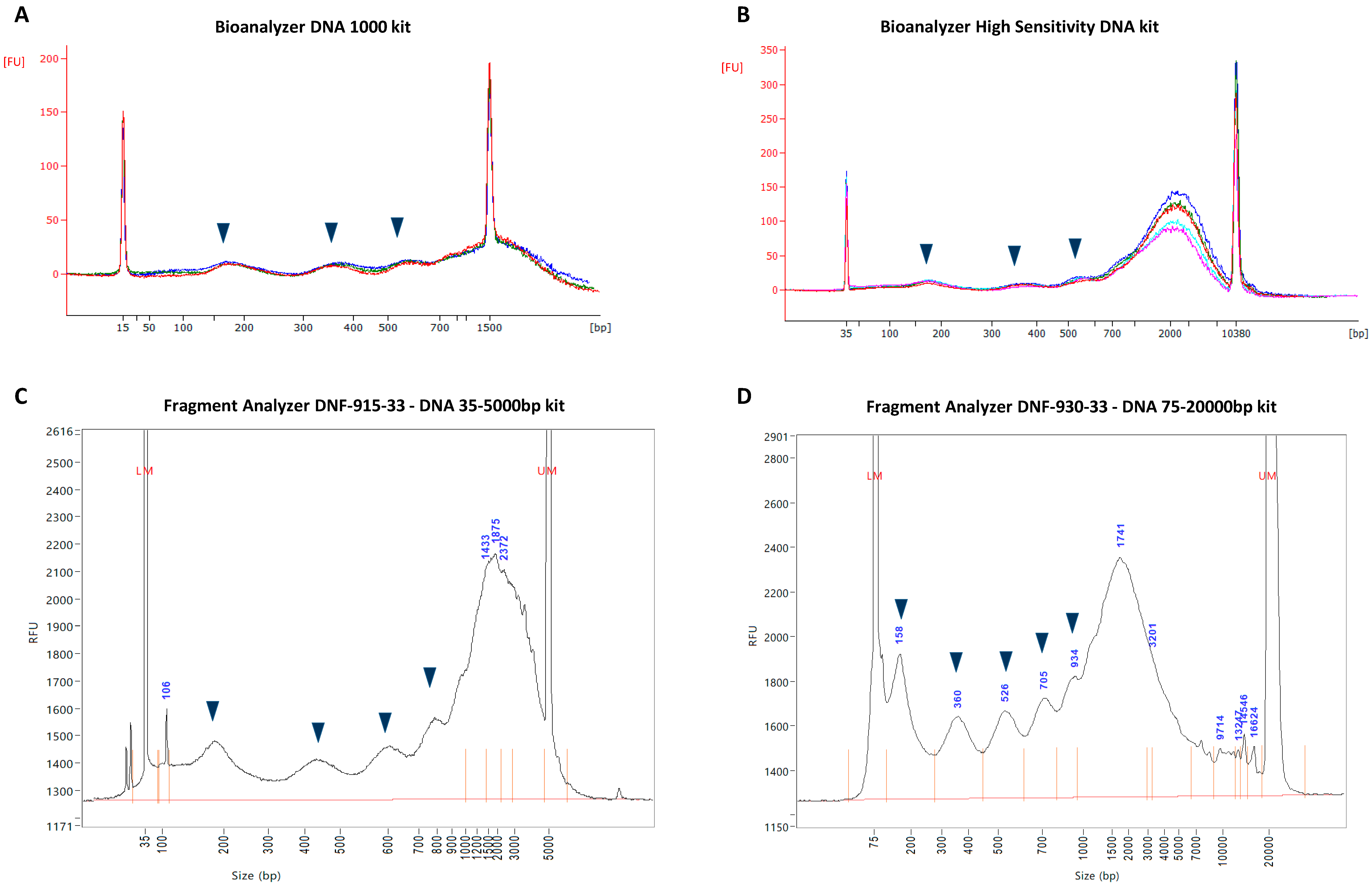
3.1. CfDNA Size Profiles as Determined by Different Microfluidic Capillary Electrophoresis Assays
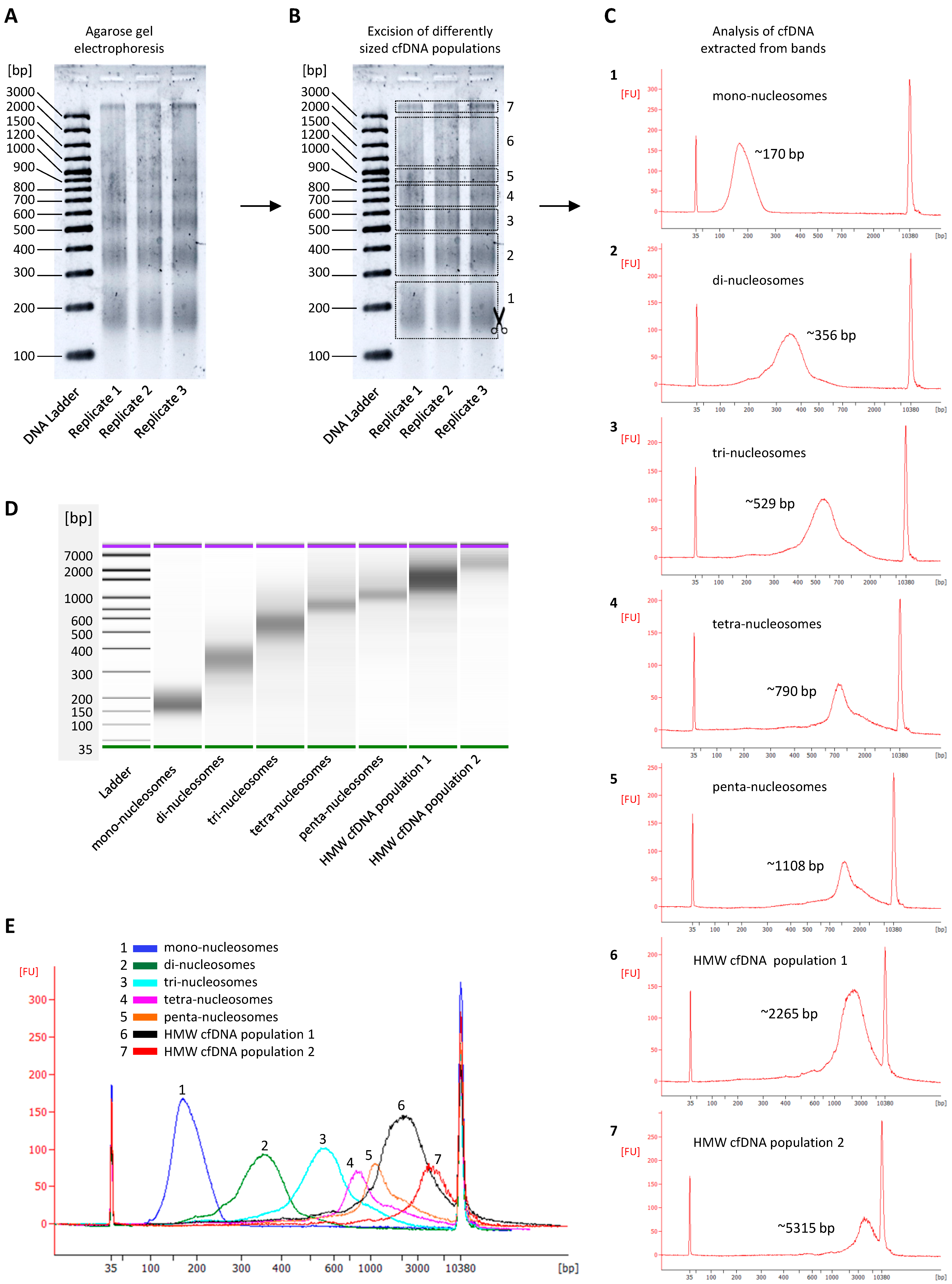
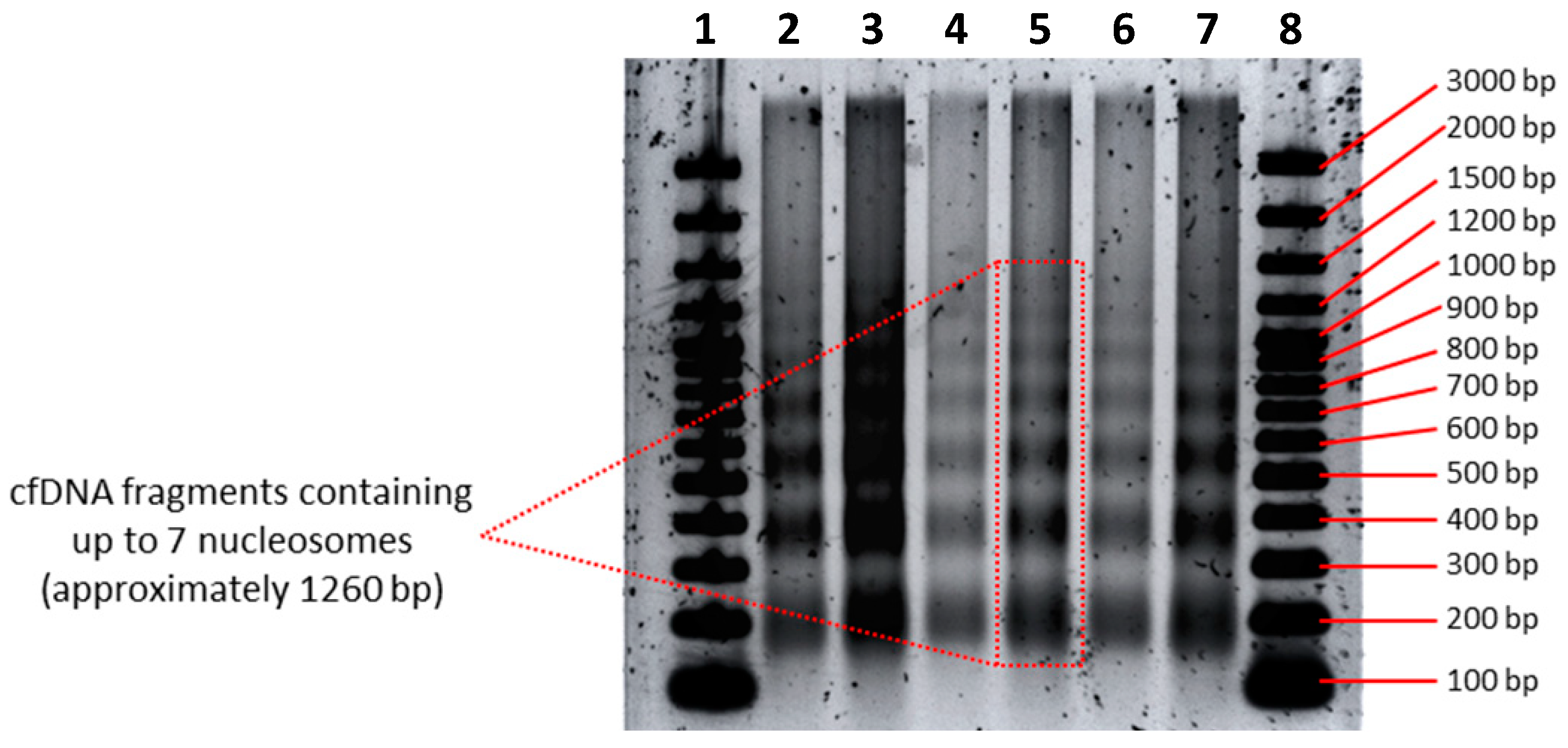
3.2. CfDNA Size Profiles as Determined by Agarose Gel Electrophoresis
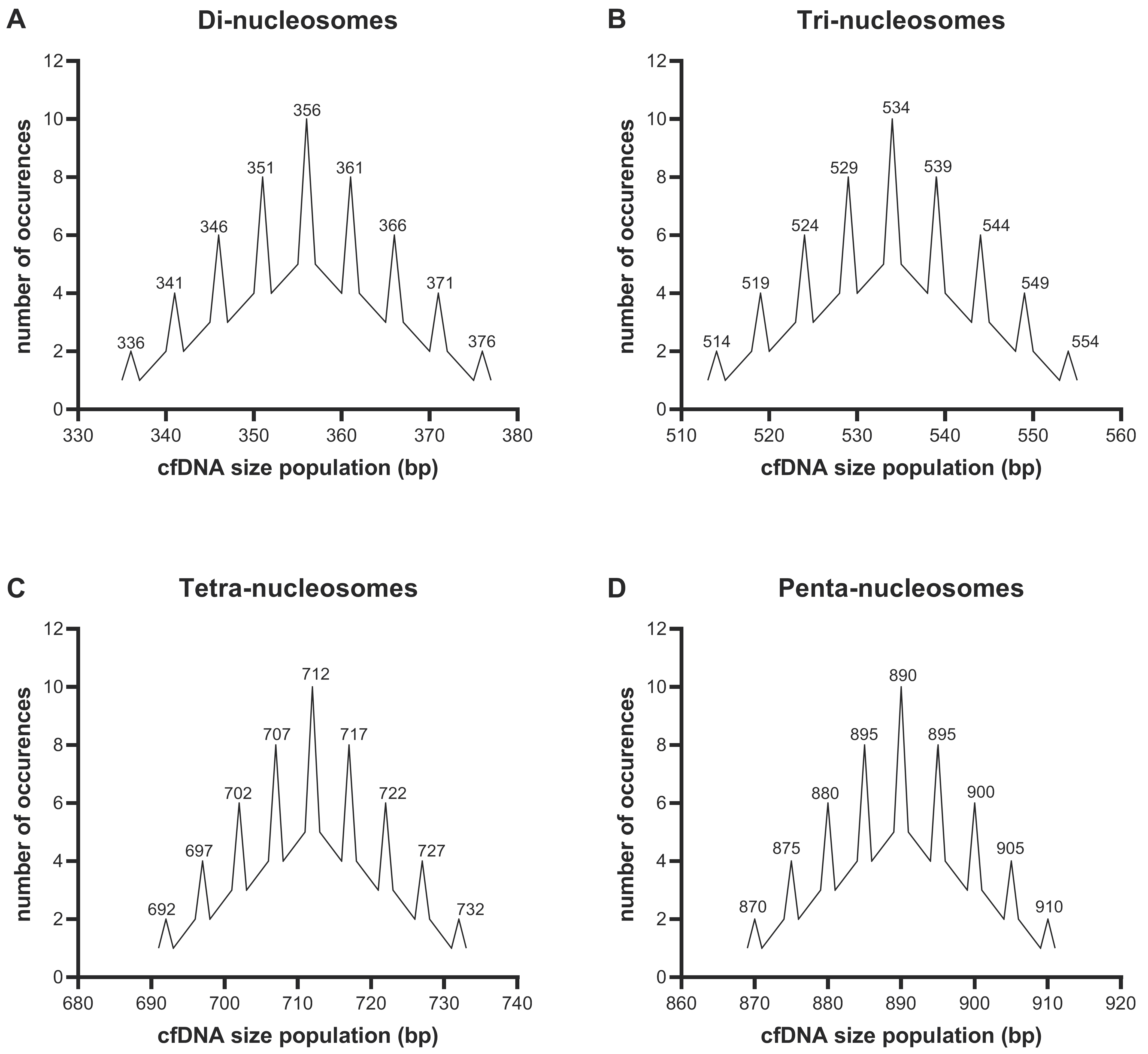
3.3. Resolution Limitations of Electrophoresis-Based cfDNA Sizing
3.4. CfDNA Degradation Patterns
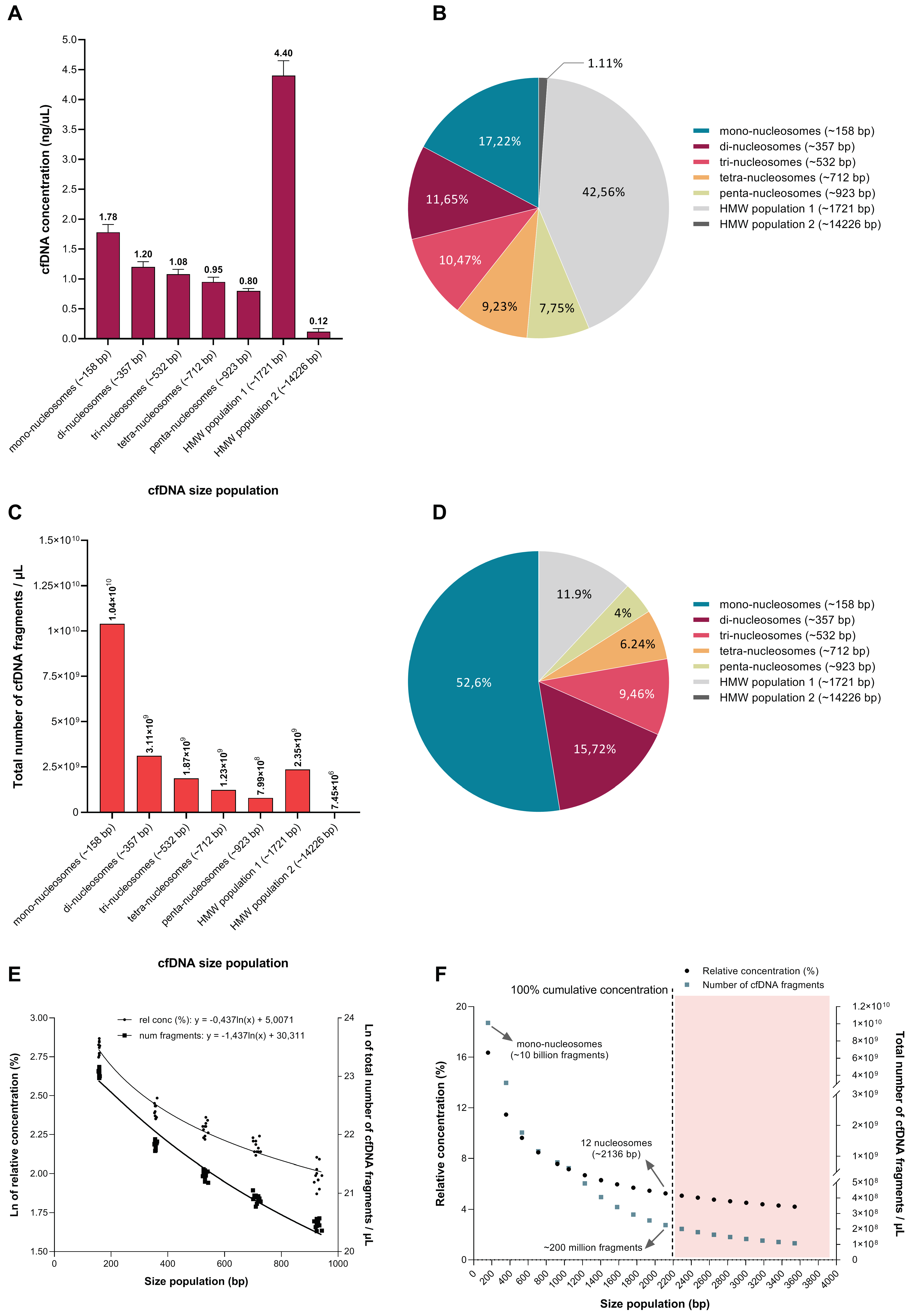
3.5. The Relative Contribution of Differently Sized cfDNA Populations
3.6. Limitations of this Study and Future Prospects
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van der Pol, Y.; Mouliere, F. Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA. Cancer Cell 2019, 36, 350–368. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Ungerer, V.; Diehl, F.; Anker, P.; Dor, Y.; Fleischhacker, M.; Gahan, P.B.; Hui, L.; Holdenrieder, S.; Thierry, A.R. Towards systematic nomenclature for cell-free DNA. Human Genet. 2021, 140, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.D.; Han, D.S.; Jiang, P.; Chiu, R.W. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 2021, 372, eaaw3616. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.C.; Lo, Y.D. Cell-Free DNA Fragmentomics in Liquid Biopsy. Diagnostics 2022, 12, 978. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef] [PubMed]
- Vessies, D.C.; Schuurbiers, M.M.; van der Noort, V.; Schouten, I.; Linders, T.C.; Lanfermeijer, M.; Ramkisoensing, K.L.; Hartemink, K.J.; Monkhorst, K.; van den Heuvel, M.M. Combining variant detection and fragment length analysis improves detection of minimal residual disease in post-surgery circulating tumour DNA of stage II-IIIA NSCLC patients. Mol. Oncol. 2022, 16, 2719–2732. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef]
- Underhill, H.R. Leveraging the fragment length of circulating tumour DNA to improve molecular profiling of solid tumour malignancies with next-generation sequencing: A pathway to advanced non-invasive diagnostics in precision oncology? Mol. Diagn. Ther. 2021, 25, 389–408. [Google Scholar] [CrossRef]
- Zviran, A.; Schulman, R.C.; Shah, M.; Hill, S.T.; Deochand, S.; Khamnei, C.C.; Maloney, D.; Patel, K.; Liao, W.; Widman, A.J. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med. 2020, 26, 1114–1124. [Google Scholar] [CrossRef]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef]
- Markus, H.; Chandrananda, D.; Moore, E.; Mouliere, F.; Morris, J.; Brenton, J.D.; Smith, C.G.; Rosenfeld, N. Refined characterization of circulating tumor DNA through biological feature integration. Sci. Rep. 2022, 12, 1928. [Google Scholar] [CrossRef]
- Chiu, R.W.; Heitzer, E.; Lo, Y.D.; Mouliere, F.; Tsui, D.W. Cell-free DNA fragmentomics: The new “Omics” on the block. Clin. Chem. 2020, 66, 1480–1484. [Google Scholar] [CrossRef]
- Sanchez, C.; Snyder, M.W.; Tanos, R.; Shendure, J.; Thierry, A.R. New insights into structural features and optimal detection of circulating tumor DNA determined by single-strand DNA analysis. NPJ Genom. Med. 2018, 3, 31. [Google Scholar] [CrossRef]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef]
- Chandrananda, D.; Thorne, N.P.; Bahlo, M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med. Genom. 2015, 8, 29. [Google Scholar] [CrossRef]
- Chan, K.A.; Jiang, P.; Sun, K.; Cheng, Y.K.; Tong, Y.K.; Cheng, S.H.; Wong, A.I.; Hudecova, I.; Leung, T.Y.; Chiu, R.W. Second generation noninvasive fetal genome analysis reveals de novo mutations, single-base parental inheritance, and preferred DNA ends. Proc. Natl. Acad. Sci. USA 2016, 113, E8159–E8168. [Google Scholar] [CrossRef]
- Sun, K.; Jiang, P.; Wong, A.I.C.; Cheng, Y.K.Y.; Cheng, S.H.; Zhang, H.; Chan, K.C.A.; Leung, T.Y.; Chiu, R.W.K.; Lo, Y.M.D. Size-tagged preferred ends in maternal plasma DNA shed light on the production mechanism and show utility in noninvasive prenatal testing. Proc. Natl. Acad. Sci. USA 2018, 115, E5106–E5114. [Google Scholar] [CrossRef]
- Jiang, P.; Xie, T.; Ding, S.C.; Zhou, Z.; Cheng, S.H.; Chan, R.W.; Lee, W.-S.; Peng, W.; Wong, J.; Wong, V.W. Detection and characterization of jagged ends of double-stranded DNA in plasma. Genome Res. 2020, 30, 1144–1153. [Google Scholar] [CrossRef]
- Jiang, P.; Sun, K.; Tong, Y.K.; Cheng, S.H.; Cheng, T.H.; Heung, M.M.; Wong, J.; Wong, V.W.; Chan, H.L.; Chan, K.A. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E10925–E10933. [Google Scholar] [CrossRef]
- Serpas, L.; Chan, R.W.Y.; Jiang, P.; Ni, M.; Sun, K.; Rashidfarrokhi, A.; Soni, C.; Sisirak, V.; Lee, W.-S.; Cheng, S.H.; et al. Dnase1l3 deletion causes aberrations in length and end-motif frequencies in plasma DNA. Proc. Natl. Acad. Sci. USA 2018, 116, 641–649. [Google Scholar] [CrossRef]
- Sun, K.; Jiang, P.; Cheng, S.H.; Cheng, T.H.T.; Wong, J.; Wong, V.W.S.; Ng, S.S.M.; Ma, B.B.Y.; Leung, T.Y.; Chan, S.L.; et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. 2019, 29, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Ulz, P.; Perakis, S.; Zhou, Q.; Moser, T.; Belic, J.; Lazzeri, I.; Wolfler, A.; Zebisch, A.; Gerger, A.; Pristauz, G.; et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat. Commun. 2019, 10, 4666. [Google Scholar] [CrossRef] [PubMed]
- Ulz, P.; Thallinger, G.G.; Auer, M.; Graf, R.; Kashofer, K.; Jahn, S.W.; Abete, L.; Pristauz, G.; Petru, E.; Geigl, J.B.; et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 2016, 48, 1273–1278. [Google Scholar] [CrossRef]
- Sin, S.T.; Jiang, P.; Deng, J.; Ji, L.; Cheng, S.H.; Dutta, A.; Leung, T.Y.; Chan, K.A.; Chiu, R.W.; Lo, Y.D. Identification and characterization of extrachromosomal circular DNA in maternal plasma. Proc. Natl. Acad. Sci. USA 2020, 117, 1658–1665. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, F.; Du, M.; Zhang, P.; Fu, S.; Wang, L. Molecular characterization of cell-free eccDNAs in human plasma. Sci. Rep. 2017, 7, 10968. [Google Scholar] [CrossRef]
- Kumar, P.; Dillon, L.W.; Shibata, Y.; Jazaeri, A.A.; Jones, D.R.; Dutta, A. Normal and Cancerous Tissues Release Extrachromosomal Circular DNA (eccDNA) into the Circulation. Mol. Cancer Res. 2017, 15, 1197–1205. [Google Scholar] [CrossRef]
- Grabuschnig, S.; Bronkhorst, A.J.; Holdenrieder, S.; Rosales Rodriguez, I.; Schliep, K.P.; Schwendenwein, D.; Ungerer, V.; Sensen, C.W. Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. Int. J. Mol. Sci. 2020, 21, 8062. [Google Scholar] [CrossRef]
- van der Pol, Y.; Moldovan, N.; Verkuijlen, S.; Ramaker, J.; Boers, D.; Onstenk, W.; de Rooij, J.; Bahce, I.; Pegtel, D.M.; Mouliere, F. The Effect of Preanalytical and Physiological Variables on Cell-Free DNA Fragmentation. Clin. Chem. 2022, 68, 803–813. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Wentzel, J.F.; Aucamp, J.; van Dyk, E.; du Plessis, L.; Pretorius, P.J. Characterization of the cell-free DNA released by cultured cancer cells. BBA-Mol Cell Res. 2016, 1863, 157–165. [Google Scholar] [CrossRef]
- Aucamp, J.; Bronkhorst, A.J.; Peters, D.L.; Van Dyk, H.C.; Van der Westhuizen, F.H.; Pretorius, P.J. Kinetic analysis, size profiling, and bioenergetic association of DNA released by selected cell lines in vitro. Cell. Mol. Life Sci. 2017, 74, 2689–2707. [Google Scholar] [CrossRef]
- Ungerer, V.; Bronkhorst, A.J.; Van den Ackerveken, P.; Herzog, M.; Holdenrieder, S. Serial profiling of cell-free DNA and nucleosome histone modifications in cell cultures. Sci. Rep. 2021, 11, 9460. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Wentzel, J.F.; Ungerer, V.; Peters, D.L.; Aucamp, J.; de Villiers, E.P.; Holdenrieder, S.; Pretorius, P.J. Sequence analysis of cell-free DNA derived from cultured human bone osteosarcoma (143B) cells. Tumour Biol. 2018, 40, 1010428318801190. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. Comparison of methods for the quantification of cell-free DNA isolated from cell culture supernatant. Tumor Biol. 2019, 41, 101042831986636. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. Comparison of methods for the isolation of cell-free DNA from cell culture supernatant. Tumor Biol. 2020, 42, 1010428320916314. [Google Scholar] [CrossRef]
- Ziraldo, R.; Shoura, M.J.; Fire, A.Z.; Levene, S.D. Deconvolution of nucleic-acid length distributions: A gel electrophoresis analysis tool and applications. Nucleic Acids Res. 2019, 47, e92. [Google Scholar] [CrossRef]
- Korolev, N.; Lyubartsev, A.P.; Nordenskiöld, L. A systematic analysis of nucleosome core particle and nucleosome-nucleosome stacking structure. Sci. Rep. 2018, 8, 1543. [Google Scholar] [CrossRef]
- Allan, J.; Fraser, R.M.; Owen-Hughes, T.; Keszenman-Pereyra, D. Micrococcal nuclease does not substantially bias nucleosome mapping. J. Mol. Biol. 2012, 417, 152–164. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, R.; Li, J.; Zhang, R. Size profile of cell-free DNA: A beacon guiding the practice and innovation of clinical testing. Theranostics 2020, 10, 4737. [Google Scholar] [CrossRef]
- Makarov, V.L.; Lejnine, S.; Bedoyan, J.; Langmore, J.P. Nucleosomal organization of telomere-specific chromatin in rat. Cell 1993, 73, 775–787. [Google Scholar] [CrossRef]
- Tremethick, D.J. Higher-order structures of chromatin: The elusive 30 nm fiber. Cell 2007, 128, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Aucamp, J.; Van Dyk, H.C.; Bronkhorst, A.J.; Pretorius, P.J. Valproic acid alters the content and function of the cell-free DNA released by hepatocellular carcinoma (HepG2) cells in vitro. Biochimie 2017, 140, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Pantazi, C.; Kolios, G.; Kakolyris, S.; Chatzaki, E. Circulating cell-free DNA release in vitro: Kinetics, size profiling, and cancer-related gene methylation. J. Cell. Physiol. 2019, 234, 14079–14089. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.C.; Jiang, P.; Peng, W.; Cheng, S.H.; Cheung, Y.T.; Tse, O.O.; Shang, H.; Poon, L.C.; Leung, T.Y.; Chan, K.A. Single-molecule sequencing reveals a large population of long cell-free DNA molecules in maternal plasma. Proc. Natl. Acad. Sci. USA 2021, 118, e2114937118. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Jiang, P.; Sun, K.; Cheng, Y.K.; Chan, K.A.; Leung, T.Y.; Chiu, R.W.; Lo, Y.D. Noninvasive prenatal testing by nanopore sequencing of maternal plasma DNA: Feasibility assessment. Clin. Chem. 2015, 61, 1305–1306. [Google Scholar] [CrossRef]
- Lang, D.; Zhang, S.; Ren, P.; Liang, F.; Sun, Z.; Meng, G.; Tan, Y.; Li, X.; Lai, Q.; Han, L. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. Gigascience 2020, 9, giaa123. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Aucamp, J.; Pretorius, P.J. Cell-free DNA: Preanalytical variables. Clin. Chim. Acta 2015, 450, 243–253. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Instrument | Kit | Separation Range (bp) | Input Concentration Range |
|---|---|---|---|
| Agilent 2100 Bioanalyzer | DNA 1000 kit | 25–1000 bp | 0.5–50 ng/µL |
| High Sensitivity (HS) DNA Kit | 50–7000 bp | 5–500 pg/µL | |
| Agilent Fragment Analyzer | dsDNA 915 Reagent kit (35–5000 bp) | 35–5000 bp | 0.5–50 ng/µL |
| dsDNA 930 Reagent kit (75–20,000 bp) | 75–20,000 bp | 0.5–50 ng/µL |
| Size Population | Biol Rep 1 (Culture Flask 1) | Biol Rep 2 (Culture Flask 2) | All Reps | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Extr Rep 1 | Extr Rep 2 | Extr Rep 1 | Extr Rep 2 | |||||||
| Avg | Stdev | Avg | Stdev | Avg | Stdev | Avg | Stdev | Avg | Stdev | |
| Mono-nucleosomes | 157.60 | 0.89 | 158.00 | 1.41 | 158.00 | 1.41 | 158.00 | 1.41 | 157.90 | 0.20 |
| Di-nucleosomes | 356.80 | 3.03 | 350.80 | 5.81 | 355.00 | 2.00 | 350.40 | 4.51 | 353.25 | 3.15 |
| Tri-nucleosomes | 532.40 | 5.13 | 529.40 | 4.22 | 532.80 | 6.83 | 528.80 | 5.76 | 530.85 | 2.04 |
| Tetra-nucleosomes | 712.40 | 7.89 | 701.00 | 5.66 | 714.20 | 10.62 | 704.00 | 8.89 | 707.90 | 6.40 |
| Penta-nucleosomes | 923.40 | 5.22 | 913.33 | 15.95 | 930.40 | 8.05 | 902.50 | 40.31 | 917.41 | 12.16 |
| HMW population 1 | 1721.00 | 14.14 | 1714.00 | 89.56 | 1736.80 | 42.99 | 1684.80 | 97.12 | 1714.15 | 21.77 |
| HMW population 2 | 14,226.24 | 2283.82 | 13,181.24 | 3196.39 | 14,034.72 | 2449.88 | 12,657.88 | 3738.73 | 13,525.02 | 735.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungerer, V.; Bronkhorst, A.J.; Uhlig, C.; Holdenrieder, S. Cell-Free DNA Fragmentation Patterns in a Cancer Cell Line. Diagnostics 2022, 12, 1896. https://doi.org/10.3390/diagnostics12081896
Ungerer V, Bronkhorst AJ, Uhlig C, Holdenrieder S. Cell-Free DNA Fragmentation Patterns in a Cancer Cell Line. Diagnostics. 2022; 12(8):1896. https://doi.org/10.3390/diagnostics12081896
Chicago/Turabian StyleUngerer, Vida, Abel J. Bronkhorst, Carsten Uhlig, and Stefan Holdenrieder. 2022. "Cell-Free DNA Fragmentation Patterns in a Cancer Cell Line" Diagnostics 12, no. 8: 1896. https://doi.org/10.3390/diagnostics12081896