
Tracing the Origin of Cell-Free DNA Molecules through Tissue-Specific Epigenetic Signatures


Abstract
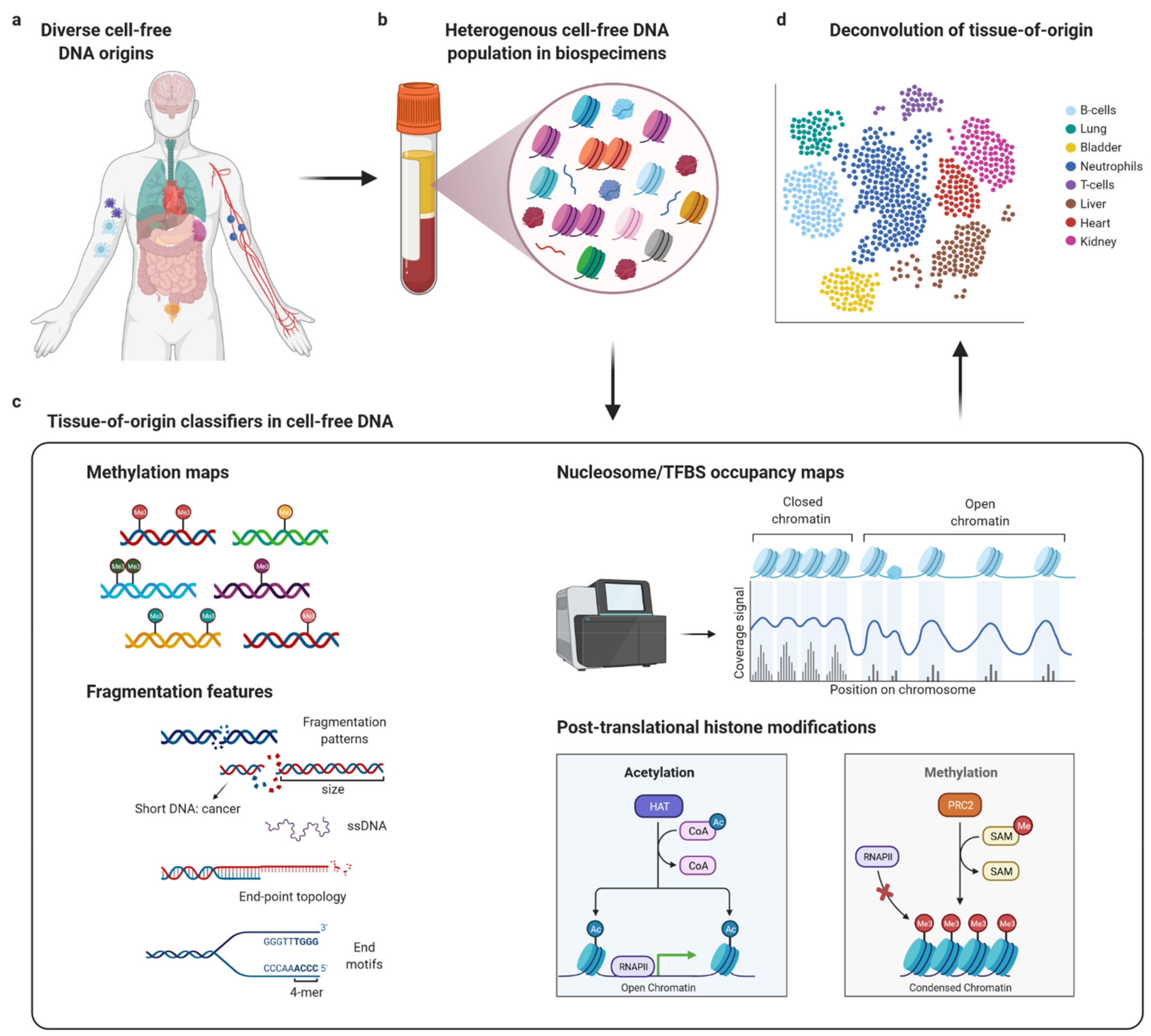
:1. Introduction
2. Epigenetic-Based Biomarkers in Liquid Biopsy
2.1. Methylation-Based Tissue-of-Origin Analysis
2.2. Nucleosome Positioning-Based Tissue-of-Origin Analysis
2.3. Tissue-of-Origin Analysis by Inferring Transcription Factor Binding Sites
2.4. Tissue-of-Origin Analysis Utilizing Histone Modifications
2.5. Tissue-of-Origin Analysis Based on Fragmentomics
2.6. Bioinformatic Analysis of Epigenetic Features of cfDNA
3. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Nawroz, H.; Koch, W.; Anker, P.; Stroun, M.; Sidransky, D. Microsatellite alterations in serum DNA of head and neck cancer patients. Nat. Med. 1996, 2, 1035–1037. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.M.; Tein, M.S.; Pang, C.C.; Yeung, C.K.; Tong, K.L.; Hjelm, N.M. Presence of donor-specific DNA in plasma of kidney and liver-transplant recipients. Lancet 1998, 351, 1329–1330. [Google Scholar] [CrossRef]
- Hui, L.; Maron, J.L.; Gahan, P.B. Other Body Fluids as Non-invasive Sources of Cell-Free DNA/RNA. In Circulating Nucleic Acids in Early Diagnosis, Prognosis and Treatment Monitoring. Advances in Predictive, Preventive and Personalised Medicine; Gahan, P., Ed.; Springer: Dordrecht, The Netherlands, 2015; Volume 5. [Google Scholar]
- Mandel, P.; Metais, P. Nuclear Acids In Human Blood Plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]
- Kohler, C.; Radpour, R.; Barekati, Z.; Asadollahi, R.; Bitzer, J.; Wight, E.; Burki, N.; Diesch, C.; Holzgreve, W.; Zhong, X.Y. Levels of plasma circulating cell free nuclear and mitochondrial DNA as potential biomarkers for breast tumors. Mol. Cancer 2009, 8, 105. [Google Scholar] [CrossRef] [Green Version]
- Zachariah, R.R.; Schmid, S.; Buerki, N.; Radpour, R.; Holzgreve, W.; Zhong, X. Levels of circulating cell-free nuclear and mitochondrial DNA in benign and malignant ovarian tumors. Obstet. Gynecol. 2008, 112, 843–850. [Google Scholar] [CrossRef]
- Kowarsky, M.; Camunas-Soler, J.; Kertesz, M.; De Vlaminck, I.; Koh, W.; Pan, W.; Martin, L.; Neff, N.F.; Okamoto, J.; Wong, R.J.; et al. Numerous uncharacterized and highly divergent microbes which colonize humans are revealed by circulating cell-free DNA. Proc. Natl. Acad. Sci. USA 2017, 114, 9623–9628. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Zhang, Y.; Gong, Y.; Sun, R.; Su, L.; Lin, X.; Shen, A.; Zhou, J.; Caiji, Z.; Wang, X.; et al. Diagnosis of Sepsis with Cell-free DNA by Next-Generation Sequencing Technology in ICU Patients. Arch. Med. Res. 2016, 47, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Grabuschnig, S.; Bronkhorst, A.J.; Holdenrieder, S.; Rosales Rodriguez, I.; Schliep, K.P.; Schwendenwein, D.; Ungerer, V.; Sensen, C.W. Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. Int. J. Mol. Sci. 2020, 21, 8062. [Google Scholar] [CrossRef]
- To, E.W.; Chan, K.C.; Leung, S.F.; Chan, L.Y.; To, K.F.; Chan, A.T.; Johnson, P.J.; Lo, Y.M. Rapid clearance of plasma Epstein-Barr virus DNA after surgical treatment of nasopharyngeal carcinoma. Clin. Cancer Res. 2003, 9, 3254–3259. [Google Scholar] [PubMed]
- Yao, W.; Mei, C.; Nan, X.; Hui, L. Evaluation and comparison of in vitro degradation kinetics of DNA in serum, urine and saliva: A qualitative study. Gene 2016, 590, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Delgado, P.O.; Alves, B.C.; de Sousa Gehrke, F.; Kuniyoshi, R.K.; Wroclavski, M.L.; Del Giglio, A.; Fonseca, F.L. Characterization of cell-free circulating DNA in plasma in patients with prostate cancer. Tumor Biol. 2013, 34, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Sai, S.; Ichikawa, D.; Tomita, H.; Ikoma, D.; Tani, N.; Ikoma, H.; Kikuchi, S.; Fujiwara, H.; Ueda, Y.; Otsuji, E. Quantification of plasma cell-free DNA in patients with gastric cancer. Anticancer Res. 2007, 27, 2747–2751. [Google Scholar]
- Lehmann-Werman, R.; Magenheim, J.; Moss, J.; Neiman, D.; Abraham, O.; Piyanzin, S.; Zemmour, H.; Fox, I.; Dor, T.; Grompe, M.; et al. Monitoring liver damage using hepatocyte-specific methylation markers in cell-free circulating DNA. JCI Insight 2018, 3, e120687. [Google Scholar] [CrossRef] [Green Version]
- Zemmour, H.; Planer, D.; Magenheim, J.; Moss, J.; Neiman, D.; Gilon, D.; Korach, A.; Glaser, B.; Shemer, R.; Landesberg, G.; et al. Non-invasive detection of human cardiomyocyte death using methylation patterns of circulating DNA. Nat. Commun. 2018, 9, 1443. [Google Scholar] [CrossRef]
- Yu, S.C.Y.; Jiang, P.; Peng, W.; Cheng, S.H.; Cheung, Y.T.T.; Tse, O.Y.O.; Shang, H.; Poon, L.C.; Leung, T.Y.; Chan, K.C.A.; et al. Single-molecule sequencing reveals a large population of long cell-free DNA molecules in maternal plasma. Proc. Natl. Acad. Sci. USA 2021, 118, e2114937118. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Jiang, P.; Chan, K.C.; Wong, J.; Cheng, Y.K.; Liang, R.H.; Chan, W.K.; Ma, E.S.; Chan, S.L.; Cheng, S.H.; et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA 2015, 112, E5503–E5512. [Google Scholar] [CrossRef] [Green Version]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgard, B.; Blennow, K.; Zetterberg, H.; et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrananda, D.; Thorne, N.P.; Bahlo, M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med. Genom. 2015, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.S.C.; Ni, M.; Chan, R.W.Y.; Chan, V.W.H.; Lui, K.O.; Chiu, R.W.K.; Lo, Y.M.D. The Biology of Cell-free DNA Fragmentation and the Roles of DNASE1, DNASE1L3, and DFFB. Am. J. Hum. Genet. 2020, 106, 202–214. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Sun, K.; Tong, Y.K.; Cheng, S.H.; Cheng, T.H.T.; Heung, M.M.S.; Wong, J.; Wong, V.W.S.; Chan, H.L.Y.; Chan, K.C.A.; et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E10925–E10933. [Google Scholar] [CrossRef] [Green Version]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.O.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef]
- Sadeh, R.; Sharkia, I.; Fialkoff, G.; Rahat, A.; Gutin, J.; Chappleboim, A.; Nitzan, M.; Fox-Fisher, I.; Neiman, D.; Meler, G.; et al. ChIP-seq of plasma cell-free nucleosomes identifies gene expression programs of the cells of origin. Nat. Biotechnol. 2021, 39, 586–598. [Google Scholar] [CrossRef]
- Ulz, P.; Perakis, S.; Zhou, Q.; Moser, T.; Belic, J.; Lazzeri, I.; Wolfler, A.; Zebisch, A.; Gerger, A.; Pristauz, G.; et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat. Commun. 2019, 10, 4666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Diep, D.; Plongthongkum, N.; Fung, H.L.; Zhang, K.; Zhang, K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat. Genet. 2017, 49, 635–642. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Li, Q.; Kang, S.; Same, M.; Zhou, Y.; Sun, C.; Liu, C.C.; Matsuoka, L.; Sher, L.; Wong, W.H.; et al. CancerDetector: Ultrasensitive and non-invasive cancer detection at the resolution of individual reads using cell-free DNA methylation sequencing data. Nucleic Acids Res. 2018, 46, e89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyfer, N.; Magenheim, J.; Peretz, A.; Cann, G.; Bredno, J.; Klochendler, A.; Fox-Fisher, I.; Shabi-Porat, S.; Hecht, M.; Pelet, T.; et al. A human DNA methylation atlas reveals principles of cell type-specific methylation and identifies thousands of cell type-specific regulatory elements. bioRxiv 2022. [Google Scholar] [CrossRef]
- Klein, E.A.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef]
- Ulz, P.; Thallinger, G.G.; Auer, M.; Graf, R.; Kashofer, K.; Jahn, S.W.; Abete, L.; Pristauz, G.; Petru, E.; Geigl, J.B.; et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 2016, 48, 1273–1278. [Google Scholar] [CrossRef]
- Sun, K.; Jiang, P.; Cheng, S.H.; Cheng, T.H.T.; Wong, J.; Wong, V.W.S.; Ng, S.S.M.; Ma, B.B.Y.; Leung, T.Y.; Chan, S.L.; et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. 2019, 29, 418–427. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Sun, K.; Peng, W.; Cheng, S.H.; Ni, M.; Yeung, P.C.; Heung, M.M.S.; Xie, T.; Shang, H.; Zhou, Z.; et al. Plasma DNA End-Motif Profiling as a Fragmentomic Marker in Cancer, Pregnancy, and Transplantation. Cancer Discov. 2020, 10, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esfahani, M.S.; Hamilton, E.G.; Mehrmohamadi, M.; Nabet, B.Y.; Alig, S.K.; King, D.A.; Steen, C.B.; Macaulay, C.W.; Schultz, A.; Nesselbush, M.C.; et al. Inferring gene expression from cell-free DNA fragmentation profiles. Nat. Biotechnol. 2022, 40, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Estecio, M.R.; Issa, J.P. Dissecting DNA hypermethylation in cancer. FEBS Lett. 2011, 585, 2078–2086. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.P.; Rand, K.N.; Molloy, P.L. Hypomethylation of repeated DNA sequences in cancer. Epigenomics 2010, 2, 245–269. [Google Scholar] [CrossRef]
- Galardi, F.; Luca, F.; Romagnoli, D.; Biagioni, C.; Moretti, E.; Biganzoli, L.; Leo, A.D.; Migliaccio, I.; Malorni, L.; Benelli, M. Cell-Free DNA-Methylation-Based Methods and Applications in Oncology. Biomolecules 2020, 10, 1677. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, S.J.; Harrison, J.; Paul, C.L.; Frommer, M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994, 22, 2990–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunau, C.; Clark, S.J.; Rosenthal, A. Bisulfite genomic sequencing: Systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001, 29, E65. [Google Scholar] [CrossRef] [PubMed]
- Miura, F.; Ito, T. Post-Bisulfite Adaptor Tagging for PCR-Free Whole-Genome Bisulfite Sequencing. Methods Mol. Biol. 2018, 1708, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Miura, F.; Shibata, Y.; Miura, M.; Sangatsuda, Y.; Hisano, O.; Araki, H.; Ito, T. Highly efficient single-stranded DNA ligation technique improves low-input whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2019, 47, e85. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Smallwood, S.A.; Lee, H.J.; Krueger, F.; Reik, W.; Kelsey, G. Genome-wide base-resolution mapping of DNA methylation in single cells using single-cell bisulfite sequencing (scBS-seq). Nat. Protoc. 2017, 12, 534–547. [Google Scholar] [CrossRef]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Vaisvila, R.; Ponnaluri, V.K.C.; Sun, Z.; Langhorst, B.W.; Saleh, L.; Guan, S.; Dai, N.; Campbell, M.A.; Sexton, B.S.; Marks, K.; et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 2021, 31, 1280–1289. [Google Scholar] [CrossRef]
- Williams, L.; Bei, Y.; Church, H.E.; Dai, N.; Dimalanta, E.T.; Ettwiller, L.M.; Evans, T.C.; Langhorst, B.W.; Borgaro, J.G.; Guan, S.; et al. Enzymatic Methyl-Seq: The Next Generation of Methylome Analysis. Available online: https://international.neb.com/tools-and-resources/feature-articles/enzymatic-methyl-seq-the-next-generation-of-methylome-analysis (accessed on 18 February 2022).
- Liu, Y.; Siejka-Zielinska, P.; Velikova, G.; Bi, Y.; Yuan, F.; Tomkova, M.; Bai, C.; Chen, L.; Schuster-Bockler, B.; Song, C.X. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 2019, 37, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Siejka-Zielinska, P.; Cheng, J.; Jackson, F.; Liu, Y.; Soonawalla, Z.; Reddy, S.; Silva, M.; Puta, L.; McCain, M.V.; Culver, E.L.; et al. Cell-free DNA TAPS provides multimodal information for early cancer detection. Sci. Adv. 2021, 7, eabh0534. [Google Scholar] [CrossRef] [PubMed]
- Taiwo, O.; Wilson, G.A.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L.M. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.Y.; Burgener, J.M.; Bratman, S.V.; De Carvalho, D.D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 2019, 14, 2749–2780. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, A.B.; Simmer, F.; Ma, K.; Kaan, A.; Zhu, J.; Stunnenberg, H.G. Whole-genome DNA methylation profiling using MethylCap-seq. Methods 2010, 52, 232–236. [Google Scholar] [CrossRef]
- Cross, S.H.; Charlton, J.A.; Nan, X.; Bird, A.P. Purification of CpG islands using a methylated DNA binding column. Nat. Genet. 1994, 6, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Soupir, A.C.; Wang, L. Cell-free DNA methylome profiling by MBD-seq with ultra-low input. Epigenetics 2022, 17, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [Green Version]
- De Koker, A.; Van Paemel, R.; De Wilde, B.; De Preter, K.; Callewaert, N. A versatile method for circulating cell-free DNA methylome profiling by reduced representation bisulfite sequencing. bioRxiv 2019. [Google Scholar] [CrossRef]
- Van Paemel, R.; De Koker, A.; Caggiano, C.; Morlion, A.; Mestdagh, P.; De Wilde, B.; Vandesompele, J.; De Preter, K. Genome-wide study of the effect of blood collection tubes on the cell-free DNA methylome. Epigenetics 2021, 16, 797–807. [Google Scholar] [CrossRef]
- Holmila, R.; Sklias, A.; Muller, D.C.; Degli Esposti, D.; Guilloreau, P.; McKay, J.; Sangrajrang, S.; Srivatanakul, P.; Hainaut, P.; Merle, P.; et al. Targeted deep sequencing of plasma circulating cell-free DNA reveals Vimentin and Fibulin 1 as potential epigenetic biomarkers for hepatocellular carcinoma. PLoS ONE 2017, 12, e0174265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widschwendter, M.; Evans, I.; Jones, A.; Ghazali, S.; Reisel, D.; Ryan, A.; Gentry-Maharaj, A.; Zikan, M.; Cibula, D.; Eichner, J.; et al. Methylation patterns in serum DNA for early identification of disseminated breast cancer. Genome Med. 2017, 9, 115. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Consortium, C. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, J.; Haan, K.; Heukamp, L.C.; Kahl, P.; Buttner, R.; Muller, S.C.; von Ruecker, A.; Bastian, P.J. CpG island hypermethylation in cell-free serum DNA identifies patients with localized prostate cancer. Prostate 2008, 68, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [Green Version]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Hodges, E.; Molaro, A.; Dos Santos, C.O.; Thekkat, P.; Song, Q.; Uren, P.J.; Park, J.; Butler, J.; Rafii, S.; McCombie, W.R.; et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol. Cell 2011, 44, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lun, F.M.; Chiu, R.W.; Sun, K.; Leung, T.Y.; Jiang, P.; Chan, K.C.; Sun, H.; Lo, Y.M. Noninvasive prenatal methylomic analysis by genomewide bisulfite sequencing of maternal plasma DNA. Clin. Chem. 2013, 59, 1583–1594. [Google Scholar] [CrossRef]
- Fernandez, A.F.; Assenov, Y.; Martin-Subero, J.I.; Balint, B.; Siebert, R.; Taniguchi, H.; Yamamoto, H.; Hidalgo, M.; Tan, A.C.; Galm, O.; et al. A DNA methylation fingerprint of 1628 human samples. Genome Res. 2012, 22, 407–419. [Google Scholar] [CrossRef] [Green Version]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujold, D.; Morais, D.A.L.; Gauthier, C.; Cote, C.; Caron, M.; Kwan, T.; Chen, K.C.; Laperle, J.; Markovits, A.N.; Pastinen, T.; et al. The International Human Epigenome Consortium Data Portal. Cell Syst. 2016, 3, 496–499.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.C.; Jiang, P.; Chan, C.W.; Sun, K.; Wong, J.; Hui, E.P.; Chan, S.L.; Chan, W.C.; Hui, D.S.; Ng, S.S.; et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 18761–18768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.H.T.; Jiang, P.; Tam, J.C.W.; Sun, X.; Lee, W.S.; Yu, S.C.Y.; Teoh, J.Y.C.; Chiu, P.K.F.; Ng, C.F.; Chow, K.M.; et al. Genomewide bisulfite sequencing reveals the origin and time-dependent fragmentation of urinary cfDNA. Clin. Biochem. 2017, 50, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W.; et al. CancerLocator: Non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol. 2017, 18, 53. [Google Scholar] [CrossRef] [Green Version]
- Karaglani, M.; Panagopoulou, M.; Baltsavia, I.; Apalaki, P.; Theodosiou, T.; Iliopoulos, I.; Tsamardinos, I.; Chatzaki, E. Tissue-Specific Methylation Biosignatures for Monitoring Diseases: An In Silico Approach. Int. J. Mol. Sci. 2022, 23, 2959. [Google Scholar] [CrossRef]
- Lubotzky, A.; Zemmour, H.; Neiman, D.; Gotkine, M.; Loyfer, N.; Piyanzin, S.; Ochana, B.L.; Lehmann-Werman, R.; Cohen, D.; Moss, J.; et al. Liquid biopsy reveals collateral tissue damage in cancer. JCI Insight 2022, 7, e153559. [Google Scholar] [CrossRef]
- Tse, O.Y.O.; Jiang, P.; Cheng, S.H.; Peng, W.; Shang, H.; Wong, J.; Chan, S.L.; Poon, L.C.Y.; Leung, T.Y.; Chan, K.C.A.; et al. Genome-wide detection of cytosine methylation by single molecule real-time sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2019768118. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Dong, Z.; Hubbell, E.; Kurtzman, K.N.; Oxnard, G.R.; Venn, O.; Melton, C.; Clarke, C.A.; Shaknovich, R.; Ma, T.; et al. Prognostic Significance of Blood-Based Multi-cancer Detection in Plasma Cell-Free DNA. Clin. Cancer Res. 2021, 27, 4221–4229. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Zheng, S.C. Cell-type deconvolution in epigenome-wide association studies: A review and recommendations. Epigenomics 2017, 9, 757–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titus, A.J.; Gallimore, R.M.; Salas, L.A.; Christensen, B.C. Cell-type deconvolution from DNA methylation: A review of recent applications. Hum. Mol. Genet. 2017, 26, R216–R224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.G.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschendorff, A.E.; Breeze, C.E.; Zheng, S.C.; Beck, S. A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinform. 2017, 18, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barefoot, M.E.; Loyfer, N.; Kiliti, A.J.; McDeed, A.P.; Kaplan, T.; Wellstein, A. Detection of Cell Types Contributing to Cancer From Circulating, Cell-Free Methylated DNA. Front. Genet. 2021, 12, 671057. [Google Scholar] [CrossRef]
- Im, Y.R.; Tsui, D.W.Y.; Diaz, L.A., Jr.; Wan, J.C.M. Next-Generation Liquid Biopsies: Embracing Data Science in Oncology. Trends Cancer 2021, 7, 283–292. [Google Scholar] [CrossRef]
- Zou, J.; Lippert, C.; Heckerman, D.; Aryee, M.; Listgarten, J. Epigenome-wide association studies without the need for cell-type composition. Nat. Methods 2014, 11, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Molitor, J.; Marsit, C.J. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics 2014, 30, 1431–1439. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, E.; Zaitlen, N.; Baran, Y.; Eng, C.; Hu, D.; Galanter, J.; Oh, S.; Burchard, E.G.; Eskin, E.; Zou, J.; et al. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat. Methods 2016, 13, 443–445. [Google Scholar] [CrossRef] [Green Version]
- Gagnon-Bartsch, J.A.; Speed, T.P. Using control genes to correct for unwanted variation in microarray data. Biostatistics 2012, 13, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Zhuang, J.; Widschwendter, M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics 2011, 27, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Repsilber, D.; Kern, S.; Telaar, A.; Walzl, G.; Black, G.F.; Selbig, J.; Parida, S.K.; Kaufmann, S.H.; Jacobsen, M. Biomarker discovery in heterogeneous tissue samples -taking the in-silico deconfounding approach. BMC Bioinform. 2010, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Song, C.X.; He, C.; Zhang, Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu. Rev. Biochem. 2014, 83, 585–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasanthakumar, A.; Godley, L.A. 5-hydroxymethylcytosine in cancer: Significance in diagnosis and therapy. Cancer Genet. 2015, 208, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Mellen, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Lu, X.; Shih, A.H.; Nie, J.; You, Q.; Xu, M.M.; Melnick, A.M.; Levine, R.L.; He, C. A Highly Sensitive and Robust Method for Genome-wide 5hmC Profiling of Rare Cell Populations. Mol. Cell 2016, 63, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Konstandin, N.; Bultmann, S.; Szwagierczak, A.; Dufour, A.; Ksienzyk, B.; Schneider, F.; Herold, T.; Mulaw, M.; Kakadia, P.M.; Schneider, S.; et al. Genomic 5-hydroxymethylcytosine levels correlate with TET2 mutations and a distinct global gene expression pattern in secondary acute myeloid leukemia. Leukemia 2011, 25, 1649–1652. [Google Scholar] [CrossRef] [Green Version]
- Globisch, D.; Munzel, M.; Muller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Bruckl, T.; Biel, M.; Carell, T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010, 5, e15367. [Google Scholar] [CrossRef] [Green Version]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.G.; Jiang, Y.; Qiu, R.; Rauch, T.A.; Wang, Y.; Schackert, G.; Krex, D.; Lu, Q.; Pfeifer, G.P. 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 2011, 71, 7360–7365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guler, G.D.; Ning, Y.; Ku, C.J.; Phillips, T.; McCarthy, E.; Ellison, C.K.; Bergamaschi, A.; Collin, F.; Lloyd, P.; Scott, A.; et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat. Commun. 2020, 11, 5270. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, X.; Lu, X.; You, L.; Song, Y.; Luo, Z.; Zhang, J.; Nie, J.; Zheng, W.; Xu, D.; et al. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res. 2017, 27, 1243–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.X.; Yin, S.; Ma, L.; Wheeler, A.; Chen, Y.; Zhang, Y.; Liu, B.; Xiong, J.; Zhang, W.; Hu, J.; et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res. 2017, 27, 1231–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schones, D.E.; Cui, K.; Cuddapah, S.; Roh, T.Y.; Barski, A.; Wang, Z.; Wei, G.; Zhao, K. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008, 132, 887–898. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Hada, A.; Sen, P.; Olufemi, L.; Hall, M.A.; Smith, B.Y.; Forth, S.; McKnight, J.N.; Patel, A.; Bowman, G.D.; et al. Dynamic regulation of transcription factors by nucleosome remodeling. eLife 2015, 4, e06249. [Google Scholar] [CrossRef]
- Segal, E.; Fondufe-Mittendorf, Y.; Chen, L.; Thastrom, A.; Field, Y.; Moore, I.K.; Wang, J.P.; Widom, J. A genomic code for nucleosome positioning. Nature 2006, 442, 772–778. [Google Scholar] [CrossRef]
- Radman-Livaja, M.; Rando, O.J. Nucleosome positioning: How is it established, and why does it matter? Dev. Biol. 2010, 339, 258–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, D.C.; Hainer, S.J. Genomic methods in profiling DNA accessibility and factor localization. Chromosome Res. 2020, 28, 69–85. [Google Scholar] [CrossRef] [Green Version]
- He, H.H.; Meyer, C.A.; Hu, S.S.; Chen, M.W.; Zang, C.; Liu, Y.; Rao, P.K.; Fei, T.; Xu, H.; Long, H.; et al. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat. Methods 2014, 11, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsompana, M.; Buck, M.J. Chromatin accessibility: A window into the genome. Epigenetics Chromatin 2014, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valouev, A.; Johnson, S.M.; Boyd, S.D.; Smith, C.L.; Fire, A.Z.; Sidow, A. Determinants of nucleosome organization in primary human cells. Nature 2011, 474, 516–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gil, A.; Riedlinger, T.; Ritter, O.; Saul, V.V.; Schmitz, M.L. Formaldehyde-assisted Isolation of Regulatory Elements to Measure Chromatin Accessibility in Mammalian Cells. J. Vis. Exp. 2018, 134, 57272. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.A. The use of multiple measurements in taxonomic problems. Ann. Eugen. 1936, 7, 179–188. [Google Scholar] [CrossRef]
- Shtumpf, M.; Piroeva, K.V.; Agrawal, S.P.; Jacob, D.R.; Teif, V.B. NucPosDB: A database of nucleosome positioning in vivo and nucleosomics of cell-free DNA. Chromosoma 2022, 131, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Koh, W.; Pan, W.; Gawad, C.; Fan, H.C.; Kerchner, G.A.; Wyss-Coray, T.; Blumenfeld, Y.J.; El-Sayed, Y.Y.; Quake, S.R. Noninvasive in vivo monitoring of tissue-specific global gene expression in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7361–7366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.C.; Lambowitz, A.M. Facile single-stranded DNA sequencing of human plasma DNA via thermostable group II intron reverse transcriptase template switching. Sci. Rep. 2017, 7, 8421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erger, F.; Norling, D.; Borchert, D.; Leenen, E.; Habbig, S.; Wiesener, M.S.; Bartram, M.P.; Wenzel, A.; Becker, C.; Toliat, M.R.; et al. cfNOMe-A single assay for comprehensive epigenetic analyses of cell-free DNA. Genome Med. 2020, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [Green Version]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- McBrian, M.A.; Behbahan, I.S.; Ferrari, R.; Su, T.; Huang, T.W.; Li, K.; Hong, C.S.; Christofk, H.R.; Vogelauer, M.; Seligson, D.B.; et al. Histone acetylation regulates intracellular pH. Mol. Cell 2013, 49, 310–321. [Google Scholar] [CrossRef] [Green Version]
- Di Cerbo, V.; Schneider, R. Cancers with wrong HATs: The impact of acetylation. Brief. Funct. Genom. 2013, 12, 231–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenhard, B.; Sandelin, A.; Carninci, P. Metazoan promoters: Emerging characteristics and insights into transcriptional regulation. Nat. Rev. Genet. 2012, 13, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, D.S.; Lis, J.T. Detecting protein-DNA interactions in vivo: Distribution of RNA polymerase on specific bacterial genes. Proc. Natl. Acad. Sci. USA 1984, 81, 4275–4279. [Google Scholar] [CrossRef] [Green Version]
- Solomon, M.J.; Varshavsky, A. Formaldehyde-mediated DNA-protein crosslinking: A probe for in vivo chromatin structures. Proc. Natl. Acad. Sci. USA 1985, 82, 6470–6474. [Google Scholar] [CrossRef] [Green Version]
- Dahl, J.A.; Jung, I.; Aanes, H.; Greggains, G.D.; Manaf, A.; Lerdrup, M.; Li, G.; Kuan, S.; Li, B.; Lee, A.Y.; et al. Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature 2016, 537, 548–552. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.P.; Turner, B.M. Immunoprecipitation of native chromatin: NChIP. Methods 2003, 31, 76–82. [Google Scholar] [CrossRef]
- Mansson, C.T.; Vad-Nielsen, J.; Meldgaard, P.; Nielsen, A.L.; Sorensen, B.S. EGFR transcription in non-small-cell lung cancer tumours can be revealed in ctDNA by cell-free chromatin immunoprecipitation (cfChIP). Mol. Oncol. 2021, 15, 2868–2876. [Google Scholar] [CrossRef]
- Chan, K.C.; Jiang, P.; Sun, K.; Cheng, Y.K.; Tong, Y.K.; Cheng, S.H.; Wong, A.I.; Hudecova, I.; Leung, T.Y.; Chiu, R.W.; et al. Second generation noninvasive fetal genome analysis reveals de novo mutations, single-base parental inheritance, and preferred DNA ends. Proc. Natl. Acad. Sci. USA 2016, 113, E8159–E8168. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Jiang, P.; Wong, A.I.C.; Cheng, Y.K.Y.; Cheng, S.H.; Zhang, H.; Chan, K.C.A.; Leung, T.Y.; Chiu, R.W.K.; Lo, Y.M.D. Size-tagged preferred ends in maternal plasma DNA shed light on the production mechanism and show utility in noninvasive prenatal testing. Proc. Natl. Acad. Sci. USA 2018, 115, E5106–E5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holdenrieder, S.; Stieber, P.; Chan, L.Y.; Geiger, S.; Kremer, A.; Nagel, D.; Lo, Y.M. Cell-free DNA in serum and plasma: Comparison of ELISA and quantitative PCR. Clin. Chem. 2005, 51, 1544–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcaide, M.; Cheung, M.; Hillman, J.; Rassekh, S.R.; Deyell, R.J.; Batist, G.; Karsan, A.; Wyatt, A.W.; Johnson, N.; Scott, D.W.; et al. Evaluating the quantity, quality and size distribution of cell-free DNA by multiplex droplet digital PCR. Sci. Rep. 2020, 10, 12564. [Google Scholar] [CrossRef]
- Augustus, E.; Van Casteren, K.; Sorber, L.; van Dam, P.; Roeyen, G.; Peeters, M.; Vorsters, A.; Wouters, A.; Raskin, J.; Rolfo, C.; et al. The art of obtaining a high yield of cell-free DNA from urine. PLoS ONE 2020, 15, e0231058. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Murillas, I.; Turner, N.C. Assessing HER2 Amplification in Plasma cfDNA. Methods Mol. Biol. 2018, 1768, 161–172. [Google Scholar] [CrossRef]
- Heider, K.; Wan, J.C.M.; Hall, J.; Belic, J.; Boyle, S.; Hudecova, I.; Gale, D.; Cooper, W.N.; Corrie, P.G.; Brenton, J.D.; et al. Detection of ctDNA from Dried Blood Spots after DNA Size Selection. Clin. Chem. 2020, 66, 697–705. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Y.; Lee, E.J.; Yoon, H.; Lee, D.H.; Kim, K.H. Comparison of Four Commercial Kits for Isolation of Urinary Cell-Free DNA and Sample Storage Conditions. Diagnostics 2020, 10, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aucamp, J.; Bronkhorst, A.J.; Peters, D.L.; Van Dyk, H.C.; Van der Westhuizen, F.H.; Pretorius, P.J. Kinetic analysis, size profiling, and bioenergetic association of DNA released by selected cell lines in vitro. Cell. Mol. Life Sci. 2017, 74, 2689–2707. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Wentzel, J.F.; Aucamp, J.; van Dyk, E.; du Plessis, L.; Pretorius, P.J. Characterization of the cell-free DNA released by cultured cancer cells. Biochim. Biophys. Acta 2016, 1863, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Bronkhorst, A.J.; Wentzel, J.F.; Ungerer, V.; Peters, D.L.; Aucamp, J.; de Villiers, E.P.; Holdenrieder, S.; Pretorius, P.J. Sequence analysis of cell-free DNA derived from cultured human bone osteosarcoma (143B) cells. Tumor Biol. 2018, 40, 1010428318801190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Pantazi, C.; Kolios, G.; Kakolyris, S.; Chatzaki, E. Circulating cell-free DNA release in vitro: Kinetics, size profiling, and cancer-related gene methylation. J. Cell. Physiol. 2019, 234, 14079–14089. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kong, P.; Ma, G.; Li, L.; Zhu, J.; Xia, T.; Xie, H.; Zhou, W.; Wang, S. Characterization of the release and biological significance of cell-free DNA from breast cancer cell lines. Oncotarget 2017, 8, 43180–43191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungerer, V.; Bronkhorst, A.J.; Van den Ackerveken, P.; Herzog, M.; Holdenrieder, S. Serial profiling of cell-free DNA and nucleosome histone modifications in cell cultures. Sci. Rep. 2021, 11, 9460. [Google Scholar] [CrossRef] [PubMed]
- Serpas, L.; Chan, R.W.Y.; Jiang, P.; Ni, M.; Sun, K.; Rashidfarrokhi, A.; Soni, C.; Sisirak, V.; Lee, W.S.; Cheng, S.H.; et al. Dnase1l3 deletion causes aberrations in length and end-motif frequencies in plasma DNA. Proc. Natl. Acad. Sci. USA 2019, 116, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, N.; Kamataki, A.; Yamaki, J.; Homma, Y. Characterization of circulating DNA in healthy human plasma. Clin. Chim. Acta 2008, 387, 55–58. [Google Scholar] [CrossRef]
- Ivanov, M.; Baranova, A.; Butler, T.; Spellman, P.; Mileyko, V. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genom. 2015, 16 (Suppl. 13), S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouliere, F.; Mair, R.; Chandrananda, D.; Marass, F.; Smith, C.G.; Su, J.; Morris, J.; Watts, C.; Brindle, K.M.; Rosenfeld, N. Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Mol. Med. 2018, 10, e9323. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, W.S.; Devlin, S.J. Locally weighted regression: An approach to regression analysis by local fitting. J. Am. Stat. Assoc. 1988, 83, 596–610. [Google Scholar] [CrossRef]
- Moldovan, N.; van der Pol, Y.; van den Ende, T.; Boers, D.; Verkuijlen, S.; Creemers, A.; Ramaker, J.; Vu, T.; Fransen, M.F.; Pegtel, M.; et al. Genome-wide cell-free DNA termini in patients with cancer. medRxiv 2021. [Google Scholar] [CrossRef]
- Burnham, P.; Kim, M.S.; Agbor-Enoh, S.; Luikart, H.; Valantine, H.A.; Khush, K.K.; De Vlaminck, I. Single-stranded DNA library preparation uncovers the origin and diversity of ultrashort cell-free DNA in plasma. Sci. Rep. 2016, 6, 27859. [Google Scholar] [CrossRef]
- Cheng, J.; Morselli, M.; Huang, W.; Heo, Y.J.; Pinheiro-Ferreira, T.; Li, F.; Wei, F.; Chia, D.; Kim, Y.; He, H.; et al. Plasma contains ultrashort single-stranded DNA in addition to nucleosomal cell-free DNA. iScience 2022, 25, 104554. [Google Scholar] [CrossRef]
- Hisano, O.; Ito, T.; Miura, F. Short single-stranded DNAs with putative non-canonical structures comprise a new class of plasma cell-free DNA. BMC Biol. 2021, 19, 225. [Google Scholar] [CrossRef]
- Hudecova, I.; Smith, C.G.; Hansel-Hertsch, R.; Chilamakuri, C.S.; Morris, J.A.; Vijayaraghavan, A.; Heider, K.; Chandrananda, D.; Cooper, W.N.; Gale, D.; et al. Characteristics, origin, and potential for cancer diagnostics of ultrashort plasma cell-free DNA. Genome Res. 2022, 32, 215–227. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Han, D.S.C.; Jiang, P.; Chiu, R.W.K. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 2021, 372, eaaw3616. [Google Scholar] [CrossRef]
- Tang, B.; Zhou, Y.; Wang, C.M.; Huang, T.H.; Jin, V.X. Integration of DNA methylation and gene transcription across nineteen cell types reveals cell type-specific and genomic region-dependent regulatory patterns. Sci. Rep. 2017, 7, 3626. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Epigenetic Feature | Method | Approach | Disease | Deconvolution Method | References |
|---|---|---|---|---|---|
| Methylation | CpG islands analysis | WGBS | HCC, NIPT, Transplant | QP | [21] |
| Methylation | Analysis of adjacent CpG sites | Bisulfite amplicon-seq | PDAC, CRC, Diabetes, Transplant, MS, TBI, IBD | Read-specific binary classification | [17,22] |
| Methylation | Methylation haplotype block analysis | scRRBS, WGBS | CRC, LCP | QP, Random forest, feature extension “haplotype blocks” | [30] |
| Methylation | Analysis of differentially methylated regions (DMRs) + ctDNA abundance | CfMeDIP-seq | PDAC, AML, lung and breast cancer, CRC, RCC, bladder cancer | Limma, binomial GLM (GLMnet) | [31] |
| Methylation | Cell-type methylation atlas | Microarray | Sepsis, islet transplantation, CRC, lung, breast, prostate cancer, CUP | NNLS | [32] |
| Methylation | CancerDetector | Microarray, WGBS | Liver cancer | Maximizing log-likelihood model (grid search) | [33] |
| Methylation | Cell-type methylation atlas Analysis of blocks of homogenously methylated CpG sites | Deep WGBS | COVID-19 | dynamically programmed probabilistic Bayes model, NNLS, wgbstools | [34] |
| Methylation | MCED test validation | Bisulfite amplicon-seq | 12 cancer types | Ensemble logistic regression, Perceptron | [35] |
| Nucleosome occupancy/ TFBS occupancy | Windowed protection score (L-WPS/S-WPS) analysis of long/short fragments | Deep WGS | Small-cell lung cancer, squamous cell lung cancer, colorectal adenocarcinoma, HCC, ductal carcinoma in situ breast cancer | Windowed approach, Fast Fourier transformation, no classification | [20] |
| Nucleosome occupancy | Nucleosome-depleted region (NDR) analysis | WGS | Breast cancer | ABSOLUTE [36], estimation of tumor purity and ploidy | [37] |
| TFBS occupancy | Accessibility score analysis | sWGS | Prostate adenocarcinoma, breast cancer, colon adenocarcinoma | Logistic regression | [29] |
| Histone modifications | Analysis of activating histone modifications | cfChIP-seq | Colorectal carcinoma, diverse liver diseases, AMI | Robust linear regression (rlm R-package) | [28] |
| Fragment size | Fragment size distribution analysis | sWGS + in vitro size selection | High-grade serous ovarian cancer | Logistic regression, random forest | [23] |
| Orientation of fragments | Orientation-aware plasma DNA fragmentation analysis (OCF) | WGS | Pregnancy, transplant, HCC, CRC, lung cancer | Nucleosome-depletion signal used to calculate OCF value, no classification | [38] |
| Fragment size | DNA evaluation of fragments for early interception (DELFI) | sWGS + genome-wide fragmentation pattern analysis | Breast, colorectal, lung, ovarian, pancreatic, gastric, and bile duct cancer | Multi-classifying approach, stochastic gradient boosting model | [27] |
| Fragment end motif | Motif diversity score (MDS) analysis using an adopted normalized Shannon entropy | WGS | HCC, pregnancy, liver transplantation, CRC, lung cancer; head and neck squamous cell, and nasopharyngeal carcinoma | SVM, logistic regression | [39] |
| Fragment size | Promoter fragmentation entropy (PFE) analysis using a modified Shannon index | Epigenetic expression inference from cfDNA-seq (EPIC-seq) | Non-small-cell lung cancer, diffuse large B cell lymphoma | Dirichlet-multinomial model, logistic regression | [40] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oberhofer, A.; Bronkhorst, A.J.; Uhlig, C.; Ungerer, V.; Holdenrieder, S. Tracing the Origin of Cell-Free DNA Molecules through Tissue-Specific Epigenetic Signatures. Diagnostics 2022, 12, 1834. https://doi.org/10.3390/diagnostics12081834
Oberhofer A, Bronkhorst AJ, Uhlig C, Ungerer V, Holdenrieder S. Tracing the Origin of Cell-Free DNA Molecules through Tissue-Specific Epigenetic Signatures. Diagnostics. 2022; 12(8):1834. https://doi.org/10.3390/diagnostics12081834
Chicago/Turabian StyleOberhofer, Angela, Abel J. Bronkhorst, Carsten Uhlig, Vida Ungerer, and Stefan Holdenrieder. 2022. "Tracing the Origin of Cell-Free DNA Molecules through Tissue-Specific Epigenetic Signatures" Diagnostics 12, no. 8: 1834. https://doi.org/10.3390/diagnostics12081834