
Cell-Free DNA for Genomic Analysis in Primary Mediastinal Large B-Cell Lymphoma

 , , , , , , , , ,
, , , , , , , , ,
Abstract
:1. Introduction
2. Methods
2.1. Patients
2.2. Histologic Review
2.3. Sample Collection and DNA Extraction
2.4. Mutational Profile and Copy Number Alterations
2.5. Statistical Analyses
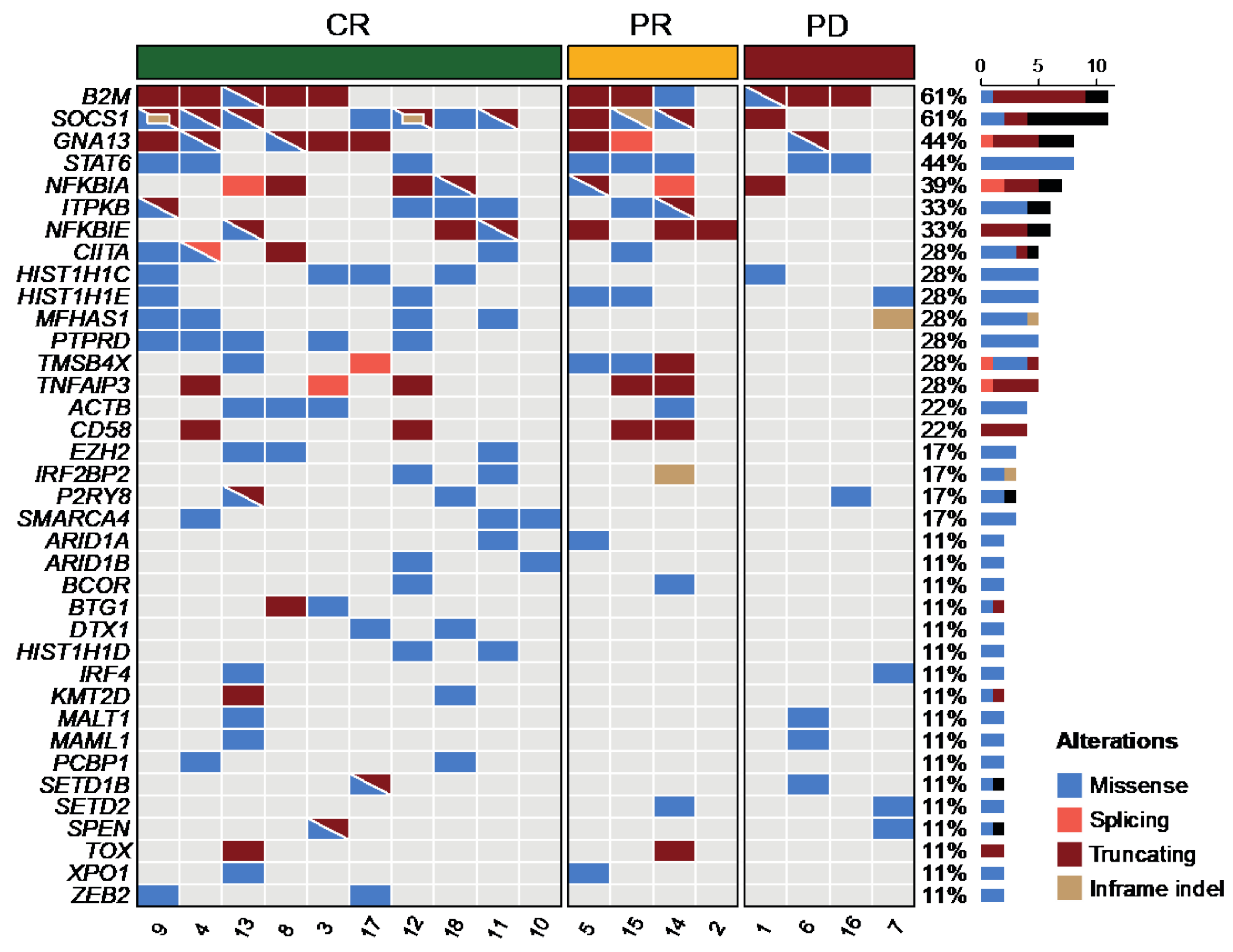
3. Results
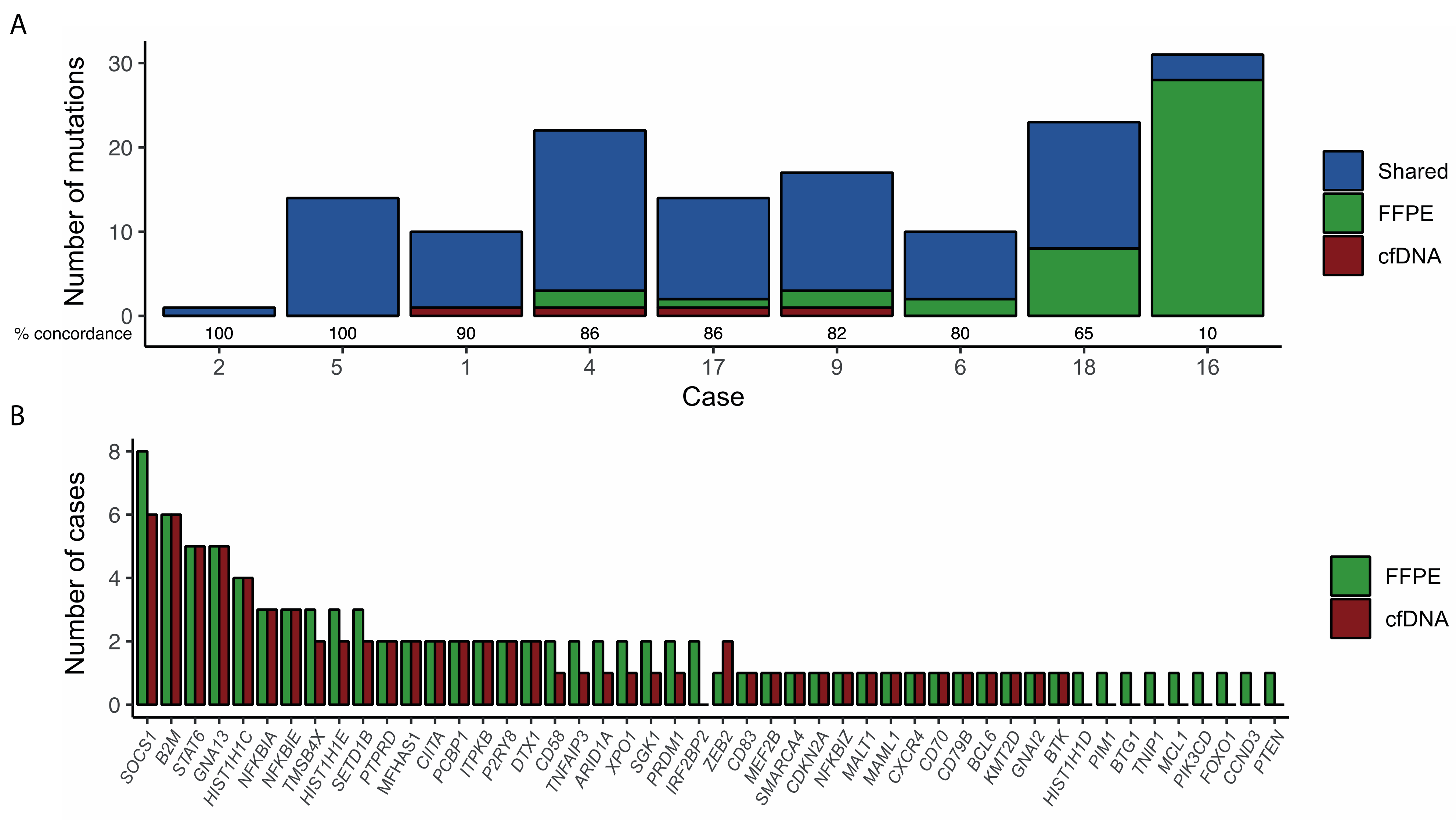
3.1. Detection of Genetic Alterations in cfDNA
3.2. Validation of Mutations in Tissue Biopsies
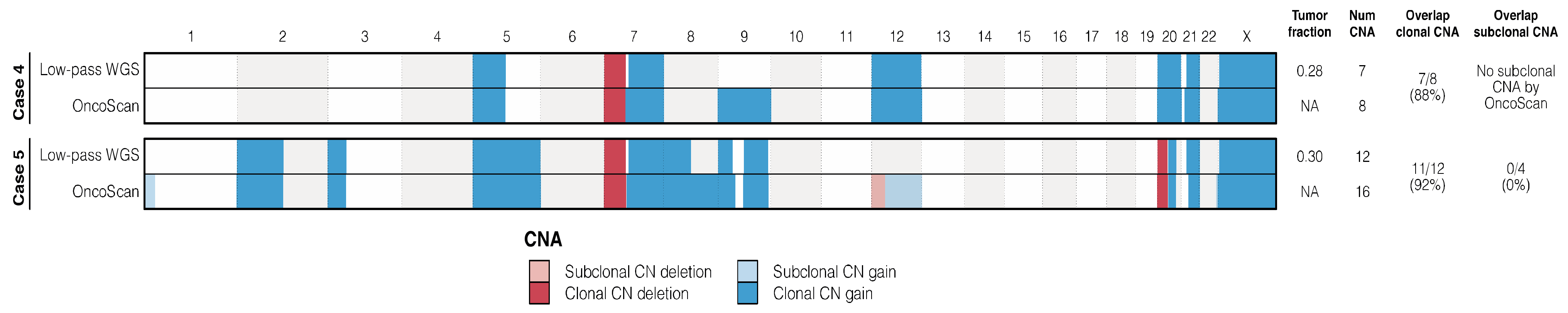
3.3. Copy Number Alterations
3.4. Tumor Burden Assessment by cfDNA
3.5. Clinical Features, Treatment, and Outcome of the Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2017. [Google Scholar]
- Sun, W.; Song, K.; Zervos, M.; Pass, H.; Cangiarella, J.; Bizekis, C.; Crawford, B.; Wang, B.Y. The diagnostic value of endobronchial ultrasound-guided needle biopsy in lung cancer and mediastinal adenopathy. Diagn. Cytopathol. 2010, 38, 337–342. [Google Scholar] [CrossRef]
- Mottok, A.; Hung, S.S.; Chavez, E.A.; Woolcock, B.; Telenius, A.; Chong, L.C.; Meissner, B.; Nakamura, H.; Rushton, C.; Viganò, E.; et al. Integrative genomic analysis identifies key pathogenic mechanisms in primary mediastinal large B-cell lymphoma. Blood 2019, 134, 802–813. [Google Scholar] [CrossRef]
- Lees, C.; Keane, C.; Gandhi, M.K.; Gunawardana, J. Biology and therapy of primary mediastinal B-cell lymphoma: Current status and future directions. Br. J. Haematol. 2019, 185, 25–41. [Google Scholar] [CrossRef]
- Lv, L.; Liu, Y. Clinical Application of Liquid Biopsy in Non-Hodgkin Lymphoma. Front. Oncol. 2021, 11, 658234. [Google Scholar] [CrossRef]
- Fernández-Lázaro, D.; Hernández, J.L.G.; García, A.C.; del Castillo, A.C.; Hueso, M.V.; Cruz-Hernández, J.J. Clinical Perspective and Translational Oncology of Liquid Biopsy. Diagnostics 2020, 10, 443. [Google Scholar] [CrossRef]
- Lakhotia, R.; Roschewski, M. Circulating tumour DNA in B-cell lymphomas: Current state and future prospects. Br. J. Haematol. 2021, 193, 867–881. [Google Scholar] [CrossRef]
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Stehr, H.; Craig, A.F.M.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transll. Med. 2016, 8, 364ra155. [Google Scholar] [CrossRef] [Green Version]
- Bohers, E.; Viailly, P.-J.; Becker, S.; Marchand, V.; Ruminy, P.; Maingonnat, C.; Bertrand, P.; Etancelin, P.; Picquenot, J.-M.; Camus, V.; et al. Non-invasive monitoring of diffuse large B-cell lymphoma by cell-free DNA high-throughput targeted sequencing: Analysis of a prospective cohort. Blood Cancer J. 2018, 8, 74. [Google Scholar] [CrossRef] [Green Version]
- Rivas-Delgado, A.; Nadeu, F.; Enjuanes, A.; Casanueva-Eliceiry, S.; Mozas, P.; Magnano, L.; de Anta, N.C.; Rovira, J.; Dlouhy, I.; Martín, S.; et al. Mutational Landscape and Tumor Burden Assessed by Cell-free DNA in Diffuse Large B-Cell Lymphoma in a Population-Based Study. Clin. Cancer Res. 2021, 27, 513–521. [Google Scholar] [CrossRef]
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C.; et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 2017, 129, 1947–1957. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Scherer, F.; Jin, M.C.; Soo, J.; Craig, A.F.M.; Esfahani, M.S.; Chabon, J.J.; Stehr, H.; Liu, C.L.; Tibshirani, R.; et al. Circulating Tumor DNA Measurements As Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2018, 36, 2845–2853. [Google Scholar] [CrossRef]
- Cheson, B.D.; Fisher, R.I.; Barrington, S.F.; Cavalli, F.; Schwartz, L.H.; Zucca, E.; Lister, T.A. Recommendations for initial evaluation, staging, and response assessment of hodgkin and non-hodgkin lymphoma: The lugano classification. J. Clin. Oncol. 2014, 32, 3059–3067. [Google Scholar] [CrossRef]
- Nadeu, F.; Delgado, J.; Royo, C.; Baumann, T.; Stankovic, T.; Pinyol, M.; Jares, P.; Navarro, A.; Martín-García, D.; Beà, S.; et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood 2016, 127, 2122–2130. [Google Scholar] [CrossRef]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [Green Version]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Camus, V.; Viennot, M.; Lévêque, E.; Viailly, P.-J.; Tonnelet, D.; Veresezan, E.-L.; Drieux, F.; Etancelin, P.; Dubois, S.; Stamatoullas, A.; et al. Circulating tumor DNA in primary mediastinal large B-cell lymphoma versus classical Hodgkin lymphoma: A retrospective study. Leuk. Lymphoma 2022, 63, 834–844. [Google Scholar] [CrossRef]
- Roschewski, M.; Phelan, J.D.; Wilson, W.H. Molecular Classification and Treatment of Diffuse Large B-Cell Lymphoma and Primary Mediastinal B-Cell Lymphoma. Cancer J. 2020, 26, 195–205. [Google Scholar] [CrossRef]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Wienand, K.; Kamburov, A.; Griffin, G.K.; Chen, P.-H.; Lako, A.; Redd, R.A.; et al. Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade. Blood 2019, 134, 2369–2382. [Google Scholar] [CrossRef]
- Rushton, C.K.; Arthur, S.E.; Alcaide, M.; Cheung, M.; Jiang, A.; Coyle, K.M.; Cleary, K.L.S.; Thomas, N.; Hilton, L.K.; Michaud, N.; et al. Genetic and evolutionary patterns of treatment resistance in relapsed B-cell lymphoma. Blood Adv. 2020, 4, 2886–2898. [Google Scholar] [CrossRef]
- Spina, V.; Khiabanian, H.; Messina, M.; Monti, S.; Cascione, L.; Bruscaggin, A.; Spaccarotella, E.; Holmes, A.B.; Arcaini, L.; Lucioni, M.; et al. The genetics of nodal marginal zone lymphoma. Blood 2016, 128, 1362–1373. [Google Scholar] [CrossRef]
- Wang, X.; Wu, B.; Yan, Z.; Wang, G.; Chen, S.; Zeng, J.; Tao, F.; Xu, B.; Ke, H.; Li, M. Association of PTPRD/PTPRT Mutation with Better Clinical Outcomes in NSCLC Patients Treated with Immune Checkpoint Blockades. Front. Oncol. 2021, 11, 650122. [Google Scholar] [CrossRef]
- Ondrejka, S.L.; Ott, G. How I Diagnose Primary Mediastinal (Thymic) Large B-Cell Lymphoma. Am. J. Clin. Pathol. 2021, 156, 497–512. [Google Scholar] [CrossRef]
- Mottok, A.; Wright, G.; Rosenwald, A.; Ott, G.; Ramsower, C.; Campo, E.; Braziel, R.M.; Delabie, J.; Weisenburger, D.D.; Song, J.Y.; et al. Molecular classification of primary mediastinal large B-cell lymphoma using routinely available tissue specimens. Blood 2018, 132, 2401–2405. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | N (%) |
|---|---|
| Median age (range) | 30 (19–68) |
| Female/Male | 11/7 (61/39) |
| ECOG-PS ≥ 2 | 2 (11) |
| B symptoms | 8 (44) |
| Stage | |
| I/II | 12 (67) |
| III/IV | 6 (33) |
| Bone marrow infiltration | 0 (0) |
| Bulky mass (>7 cm) | 14 (78) |
| Lactate dehydrogenase > normal | 14 (78) |
| IPI | |
| Low risk | 10 (56) |
| Low-Intermediate risk | 5 (28) |
| High-Intermediate risk | 2 (11) |
| High risk | 1 (5) |
| Treatment | |
| R-CHOP | 13 (72) |
| DA-R-EPOCH | 5 (28) |
| Consolidative radiotherapy | 10 (56) |
| Response to treatment | |
| Complete response | 10 (56) |
| Partial response | 4 (22) |
| Progressive disease | 4 (22) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivas-Delgado, A.; Nadeu, F.; Andrade-Campos, M.; López, C.; Enjuanes, A.; Mozas, P.; Frigola, G.; Colomo, L.; Sanchez-Gonzalez, B.; Villamor, N.; et al. Cell-Free DNA for Genomic Analysis in Primary Mediastinal Large B-Cell Lymphoma. Diagnostics 2022, 12, 1575. https://doi.org/10.3390/diagnostics12071575
Rivas-Delgado A, Nadeu F, Andrade-Campos M, López C, Enjuanes A, Mozas P, Frigola G, Colomo L, Sanchez-Gonzalez B, Villamor N, et al. Cell-Free DNA for Genomic Analysis in Primary Mediastinal Large B-Cell Lymphoma. Diagnostics. 2022; 12(7):1575. https://doi.org/10.3390/diagnostics12071575
Chicago/Turabian StyleRivas-Delgado, Alfredo, Ferran Nadeu, Marcio Andrade-Campos, Cristina López, Anna Enjuanes, Pablo Mozas, Gerard Frigola, Luis Colomo, Blanca Sanchez-Gonzalez, Neus Villamor, and et al. 2022. "Cell-Free DNA for Genomic Analysis in Primary Mediastinal Large B-Cell Lymphoma" Diagnostics 12, no. 7: 1575. https://doi.org/10.3390/diagnostics12071575