
Unusual N Gene Dropout and Ct Value Shift in Commercial Multiplex PCR Assays Caused by Mutated SARS-CoV-2 Strain

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and RT-qPCR
2.2. Whole-Genome Sequencing
2.3. Statistical Analysis
3. Results
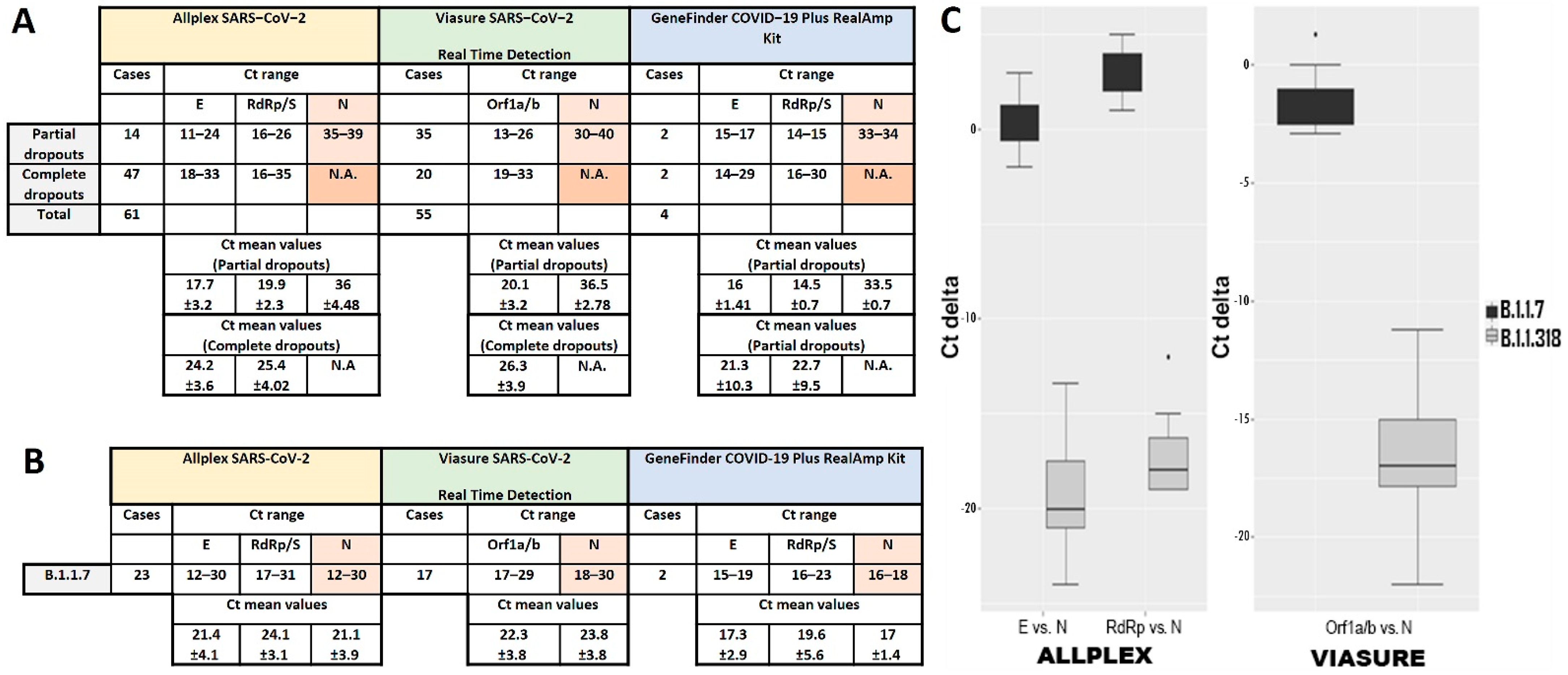
3.1. N Gene Dropout in RT-qPCR
3.2. Definition of N Gene Dropout
3.3. N Gene Amplification Delay Is Specific for the B.1.1.318 Variant
4. Discussion

{kind=link}
{kind=link}
| Variant | Gene | Detection Kit | References |
|---|---|---|---|
| B.1.1.7 | S dropout or delay | TaqPath COVID-19 CE-IVD RT-PCR Kit | [6,7,9,10] |
| Helix® COVID-19 Test | [8] | ||
| RdRp/S double curve or low amplification | Allplex SARS-CoV-2 Assay | [33] | |
| N dropout or delay | TaqPath COVID-19 CE-IVD RT-PCR Kit | [12] | |
| Allplex SARS-CoV-2/FluA/FluB/RSV Assay | [11,12] | ||
| B.1.258 | S dropout or delay | TaqPath COVID-19 CE-IVD RT-PCR Kit | [6] |
| B.1.617.2 | RdRp/S double curve or low amplification | Allplex SARS-CoV-2 Assay | [33] |
| B.1.177 (B.1.177.75) | N dropout | TaqPath COVID-19 CE-IVD RT-PCR Kit | [5] |
| B.1.1.529 | S dropout | TaqPath COVID-19 Combo Kit | [34] |
| B.1.1.318 | N dropout | Amplidiag COVID-19 | [19] |
| PerkinElmer SARS-CoV-2 Real-time RT-PCR Assay | [19] | ||
| N dropout or delay | Allplex SARS-CoV-2 | This work | |
| VIASURE SARS-CoV-2 Real Time Detection Kit | This work | ||
| GeneFinder COVID-19 Plus RealAmp Kit | This work | ||
| P.1 (or P.1.1) | N dropout | GeneFinder COVID-19 Plus RealAmp Kit | [21] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Datta, M.; Singh, D.D.; Naqvi, R.A. Molecular Diagnostic Tools for the Detection of SARS-CoV-2. Int. Rev. Immunol. 2021, 40, 143–156. [Google Scholar] [CrossRef]
- Aguiar, E.; Navas, J.; Pacheco, L.G.C. The COVID-19 Diagnostic Technology Landscape: Efficient Data Sharing Drives Diagnostic Development. Front. Public Health 2020, 8, 309. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.J.; Garner, L.V.; Smoot, J.W.; Li, Y.; Zhou, Q.; Saveson, C.J.; Sasso, J.M.; Gregg, A.C.; Soares, D.J.; Beskid, T.R.; et al. Assay Techniques and Test Development for COVID-19 Diagnosis. ACS Cent. Sci. 2020, 6, 591–605. [Google Scholar] [CrossRef]
- Decaro, N.; Lorusso, A. Novel human coronavirus (SARS-CoV-2): A lesson from animal coronaviruses. Vet. Microbiol. 2020, 244, 108693. [Google Scholar] [CrossRef]
- Amato, L.; Jurisic, L.; Puglia, I.; Di Lollo, V.; Curini, V.; Torzi, G.; Di Girolamo, A.; Mangone, I.; Mancinelli, A.; Decaro, N.; et al. Multiple detection and spread of novel strains of the SARS-CoV-2 B.1.177 (B.1.177.75) lineage that test negative by a commercially available nucleocapsid gene real-time RT-PCR. Emerg. Microbes. Infect. 2021, 10, 1148–1155. [Google Scholar] [CrossRef]
- Guerra-Assunção, J.A.; Randell, P.A.; Boshier, F.A.T.; Crone, M.A.; Pang, J.; Mahungu, T.; Freemont, P.S.; Breuer, J. Reliability of Spike Gene Target Failure for ascertaining SARS-CoV-2 lineage B.1.1.7 prevalence in a hospital setting. MedRxiv 2021, 21255084. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. MedRxiv 2020, 20249034. [Google Scholar] [CrossRef]
- Washington, N.L.; White, S.; Schiabor Barrett, K.M.; Cirulli, E.T.; Bolze, A.; Lu, J.T. S gene dropout patterns in SARS-CoV-2 tests suggest spread of the H69del/V70del mutation in the US. MedRxiv 2020. [Google Scholar] [CrossRef]
- Bal, A.; Destras, G.; Gaymard, A.; Steic, K.; Marlet, J.; Eymieux, S.; Regue, H.; Semanas, Q.; D’Aubarede, C.; Billaud, G.; et al. Two-step strategy for the identiication of SARS-CoV-2 variant of concern 202012/01 and other variants with spike deletion H69–V70, France, August to December 2020. Eurosurveillance 2021, 26, 2100008. [Google Scholar] [CrossRef]
- Buenestado-Serrano, S.; Recio, R.; Sola Campoy, P.J.; Catalán, P.; Folgueira, M.D.; Villa, J.; Gallego, I.M.; de la Cueva, V.M.; Meléndez, M.A.; Zayas, C.A.; et al. First confirmation of importation and transmission in Spain of the newly identified SARS-CoV-2 B.1.1.7 variant. Enferm. Infecc. Microbiol. Clin. 2021. [Google Scholar] [CrossRef]
- Wollschläger, P.; Todt, D.; Gerlitz, N.; Pfaender, S.; Bollinger, T.; Sing, A.; Dangel, A.; Ackermann, N.; Korn, K.; Ensser, A.; et al. SARS-CoV-2 N gene dropout and N gene Ct value shift as indicator for the presence of B.1.1.7 lineage in a commercial multiplex PCR assay. Clin. Microbiol. Infect. 2021, 9, 1353.e1–1353.e5. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Calvo, J.M.; Alados Arboledas, J.C.; Ros Vidal, L.; de Francisco, J.L.; López Prieto, M.D. Diagnostic pre-screening method based on N-gene dropout or delay to increase feasibility of SARS-CoV-2 VOC B.1.1.7 detection. Diagn. Microbiol. Infect. Dis. 2021, 4, 115491. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, L.; Sakthivel, S.K.; Whitaker, B.; Murray, J.; Kamili, S.; Lynch, B.; Malapati, L.; Burke, S.A.; Harcourt, J.; et al. US CDC Real-Time Reverse Transcription PCR Panel for Detection of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1654. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Peñaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling Fu, J.Y.; Chong, Y.M.; Sam, I.C.; Chan, Y.F. SARS-CoV-2 multiplex RT-PCR to detect variants of concern (VOCs) in Malaysia, between January to May 2021. J. Virol. Methods 2022, 10, 114462. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control (ECDC). Methods for the Detection and Characterization of SARS-CoV-2 Variants—First Update. Available online: https://www.ecdc.Europa.eu/sites/default/files/documents/Methods-for-the-detection-and-characterization-of-SARS-CoV-2-variants-first-update.pdf (accessed on 20 December 2021).
- Neopane, P.; Nypaver, J.; Shrestha, R.; Beqaj, S.S. SARS-CoV-2 Variants Detection Using TaqMan SARS-CoV-2 Mutation Panel Molecular Genotyping Assays. Infect. Drug. Resist. 2021, 14, 4471–4479. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern. Available online: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern (accessed on 26 November 2021).
- Laine, P.; Nihtilä, H.; Mustanoja, E.; Lyyski, A.; Ylinen, A.; Hurme, J.; Paulin, L.; Jokiranta, S.; Auvinen, P.; Meri, T. SARS-CoV-2 variant with mutations in N gene affecting detection by widely used PCR primers. J. Med. Virol. 2021, 2, 10. [Google Scholar] [CrossRef]
- Manouana, G.P.; Maloum, M.N.; Bikangui, R.; Bingono, S.O.O.; Nguema, G.O.; Honkpehedji, J.Y.; Rossatanga, E.G.; Zoa-Assoumou, S.; Pallerla, S.R.; Rachakonda, S.; et al. Emergence of B.1.1.318 SARS-CoV-2 viral lineage and high incidence of alpha B.1.1.7 variant of concern in the Republic of Gabon. Int. J. Infect. Dis. 2022, 114, 151–154. [Google Scholar] [CrossRef]
- Lesbon, J.C.C.; Poleti, M.D.; Oliveira, E.C.D.M.; Patané, J.S.L.; Clemente, L.G.; Viala, V.L.; Ribeiro, G.; Giovanetti, M.; de Alcantara, L.C.J.; de Lima, L.P.O.; et al. Nucleocapsid (N) Gene Mutations of SARS-CoV-2 Can Affect Real-Time RT-PCR Diagnostic and Impact False-Negative Results. Viruses 2021, 13, 2474. [Google Scholar] [CrossRef]
- Dicker, D.; Lev, S.; Gottesman, T.; Kournos, T.; Dotan, M.; Ashorov, N.; Marcoviciu, D.; Golan, R. A Time Frame for Testing Negative for SARS-COV2 in People with Obesity. Obes. Facts 2020, 13, 528–533. [Google Scholar] [CrossRef]
- Benrahma, H.; Idrissa, D.; Imane, S.; Jalila, R.; Meskaouni, N.; Benmessaoud, R.; Arouro, K.; Jaras, K.; Adam, Z.; Nahir, S.; et al. Epidemiological description and analysis of RdRp, E and N genes dynamic by RT-PCR of SARS-CoV-2 in Moroccan population: Experience of the National Reference Laboratory (LNR)-UM6SS. MedRxiv 2020, 20135137. [Google Scholar] [CrossRef]
- Falasca, F.; Sciandra, I.; Di Carlo, D.; Gentile, M.; Deales, A.; Antonelli, G.; Turriziani, O. Detection of SARS-COV N2 Gene: Very low amounts of viral RNA or false positive? J. Clin. Virol. 2020, 133, 104660. [Google Scholar] [CrossRef]
- Jaeger, L.H.; Nascimento, T.C.; Rocha, F.D.; Vilela, F.M.P.; Duque, A.P.D.N.; Silva, L.M.; Riani, L.R.; Moreira, J.P.; Chagas, J.M.D.A.; Pereira, T.V.; et al. Adjusting RT-qPCR conditions to avoid unspecific amplification in SARS-CoV-2 diagnosis. Int. J Infect. Dis. 2021, 102, 437–439. [Google Scholar] [CrossRef]
- Konrad, R.; Eberle, U.; Dangel, A.; Treis, B.; Berger, A.; Bengs, K.; Fingerle, V.; Liebl, B.; Ackermann, N.; Sing, A. Rapid establishment of laboratory diagnostics for the novel coronavirus SARS-CoV-2 in Bavaria, Germany, February 2020. Eurosurveillance 2020, 25, 2000173. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Xing, N.; Meng, K.; Fu, B.; Xue, W.; Dong, P.; Tang, W.; Xiao, Y.; Liu, G.; Luo, H.; et al. Nucleocapsid mutations R203K/G204R increase the infectivity, fitness, and virulence of SARS-CoV-2. Cell Host Microbe. 2021, 29, 1788–1801.e6. [Google Scholar] [CrossRef]
- Nagy, Á.; Pongor, S.; Győrffy, B. Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int. J. Antimicrob. Agents. 2021, 57, 106272. [Google Scholar] [CrossRef]
- Klink, G.V.; Safina, K.R.; Garushyants, S.K.; Moldovan, M.; Nabieva, E.; The CoRGI (Coronavirus Russian Genetic Initiative) Consortium; Komissarov, A.B.; Lioznov, D.; Bazykin, G.A. Spread of endemic SARS-CoV-2 lineages in Russia. MedRxiv 2021, 21257695. [Google Scholar] [CrossRef]
- Del Vecchio, C.; Brancaccio, G.; Brazzale, A.R.; Lavezzo, E.; Onelia, F.; Franchin, E.; Manuto, L.; Bianca, F.; Cianci, V.; Cattelan, A.; et al. Emergence of N antigen SARS-CoV-2 genetic variants escaping detection of antigenic tests. MedRxiv 2021, 21253802. [Google Scholar] [CrossRef]
- Kiryanov, S.A.; Levina, T.A.; Konopleva, M.V.; Suslov, A.P. Identification of Hotspot Mutations in the N Gene of SARS-CoV-2 in Russian Clinical Samples That May Affect the Detection by Reverse Transcription-PCR. Diagnostics 2022, 12, 147. [Google Scholar] [CrossRef]
- Brejová, B.; Boršová, K.; Hodorová, V.; Čabanová, V.; Reizigová, L.; Paul, E.D.; Čekan, P.; Klempa, B.; Nosek, J.; Vinař, T. A SARS-CoV-2 mutant from B.1.258 lineage with ∆H69/∆V70 deletion in the Spike protein circulating in Central Europe in the fall 2020. Virus Genes 2021, 57, 556–560. [Google Scholar] [CrossRef]
- So, M.K.; Park, S.; Lee, K.; Kim, S.K.; Chung, H.S.; Lee, M. Variant Prediction by Analyzing RdRp/S Gene Double or Low Amplification Pattern in Allplex SARS-CoV-2 Assay. Diagnostics 2021, 11, 1854. [Google Scholar] [CrossRef] [PubMed]
- Metzger, C.M.J.A.; Lienhard, R.; Seth-Smith, H.M.B.; Roloff, T.; Wegner, F.; Sieber, J.; Bel, M.; Greub, G.; Egli, A. PCR performance in the SARS-CoV-2 Omicron variant of concern? Swiss. Med. Wkly. 2021, 151, w30120. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozidis, P.; Tsaousi, E.T.; Kostoulas, C.; Sakaloglou, P.; Gouni, A.; Koumpouli, D.; Sakkas, H.; Georgiou, I.; Gartzonika, K. Unusual N Gene Dropout and Ct Value Shift in Commercial Multiplex PCR Assays Caused by Mutated SARS-CoV-2 Strain. Diagnostics 2022, 12, 973. https://doi.org/10.3390/diagnostics12040973
Bozidis P, Tsaousi ET, Kostoulas C, Sakaloglou P, Gouni A, Koumpouli D, Sakkas H, Georgiou I, Gartzonika K. Unusual N Gene Dropout and Ct Value Shift in Commercial Multiplex PCR Assays Caused by Mutated SARS-CoV-2 Strain. Diagnostics. 2022; 12(4):973. https://doi.org/10.3390/diagnostics12040973
Chicago/Turabian StyleBozidis, Petros, Eleni T. Tsaousi, Charilaos Kostoulas, Prodromos Sakaloglou, Athanasia Gouni, Despoina Koumpouli, Hercules Sakkas, Ioannis Georgiou, and Konstantina Gartzonika. 2022. "Unusual N Gene Dropout and Ct Value Shift in Commercial Multiplex PCR Assays Caused by Mutated SARS-CoV-2 Strain" Diagnostics 12, no. 4: 973. https://doi.org/10.3390/diagnostics12040973