
Liquid Profiling for Cancer Patient Stratification in Precision Medicine—Current Status and Challenges for Successful Implementation in Standard Care


Abstract
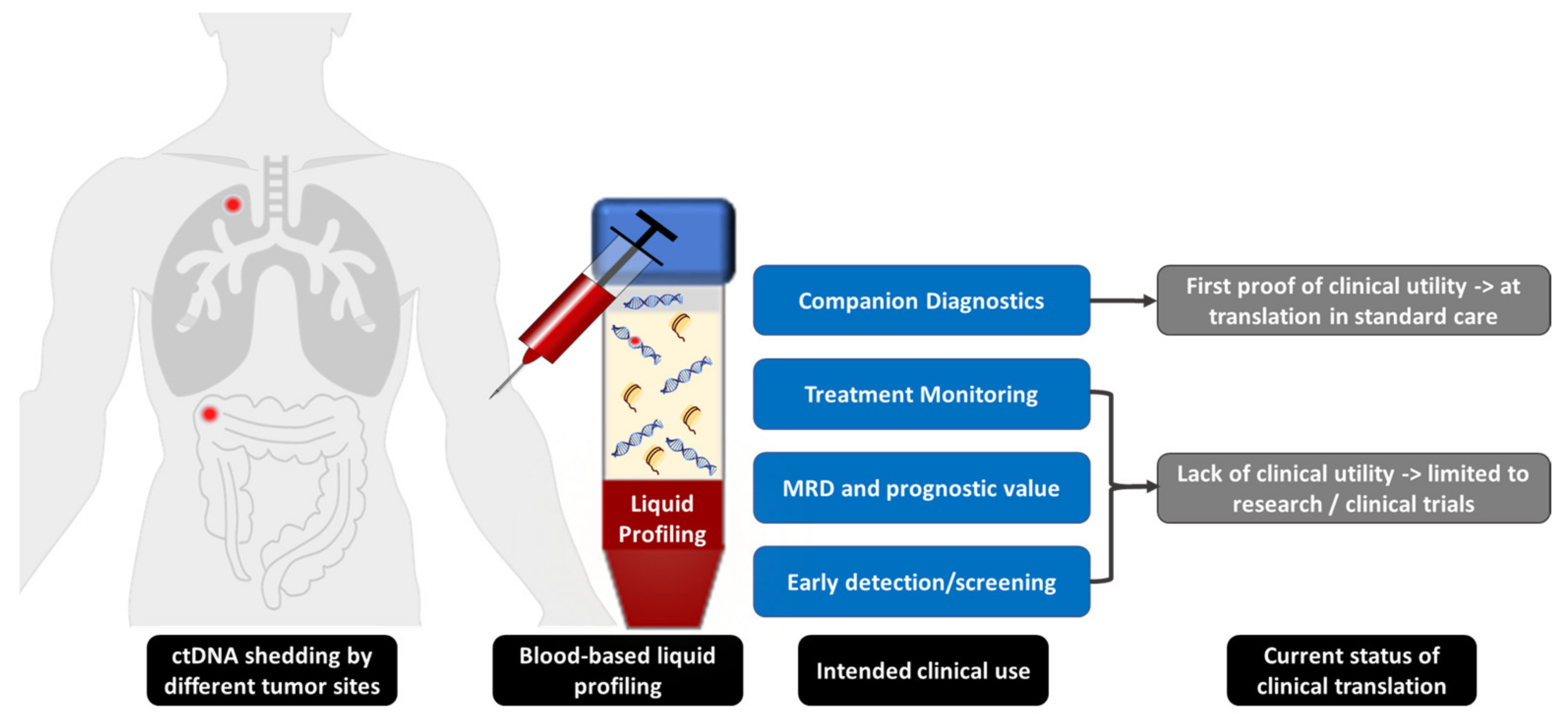
:1. Introduction
2. Clinical Application
2.1. Companion Diagnostics and Detection of Resistance Mechanisms
2.2. Treatment Monitoring
2.3. Minimal Residual Disease and Assessment of Prognosis
2.4. Early Detection/Screening
3. Current Status of and Challenges for Clinical Implementation
3.1. Technical Challenges
3.2. Interpretation of Results and Reporting
3.3. Quality Control
3.4. Clinical Acceptance
3.5. Reimbursement
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alix-Panabières, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, PO.17.00011. [Google Scholar] [CrossRef] [PubMed]
- Volckmar, A.-L.; Sültmann, H.; Riediger, A.; Fioretos, T.; Schirmacher, P.; Endris, V.; Stenzinger, A.; Dietz, S. A field guide for cancer diagnostics using cell-free DNA: From principles to practice and clinical applications. Genes Chromosom. Cancer 2018, 57, 123–139. [Google Scholar] [CrossRef]
- Overman, M.J.; Modak, J.; Kopetz, S.; Murthy, R.; Yao, J.C.; Hicks, M.E.; Abbruzzese, J.L.; Tam, A.L. Use of Research Biopsies in Clinical Trials: Are Risks and Benefits Adequately Discussed? J. Clin. Oncol. 2013, 31, 17–22. [Google Scholar] [CrossRef]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- Bonanno, L.; Pavan, A.; Ferro, A.; Calvetti, L.; Frega, S.; Pasello, G.; Aprile, G.; Guarneri, V.; Conte, P.; Rete Oncologica, V. Clinical Impact of Plasma and Tissue Next-Generation Sequencing in Advanced Non-Small Cell Lung Cancer: A Real-World Experience. Oncologist 2020, 25, e1996–e2005. [Google Scholar] [CrossRef]
- Moorcraft, S.Y.; Gonzalez, D.; Walker, B.A. Understanding next generation sequencing in oncology: A guide for oncologists. Crit. Rev. Oncol./Hematol. 2015, 96, 463–474. [Google Scholar] [CrossRef]
- Zhang, P.; Lehmann, B.D.; Shyr, Y.; Guo, Y. The Utilization of Formalin Fixed-Paraffin-Embedded Specimens in High Throughput Genomic Studies. Int. J. Genom. 2017, 2017, 1926304. [Google Scholar] [CrossRef] [PubMed]
- Gárcia, J.; Dusserre, E.; Cheynet, V.; Bringuier, P.P.; Brengle-Pesce, K.; Wozny, A.-S.; Rodriguez-Lafrasse, C.; Freyer, G.; Brevet, M.; Payen, L.; et al. Evaluation of pre-analytical conditions and comparison of the performance of several digital PCR assays for the detection of major EGFR mutations in circulating DNA from non-small cell lung cancers: The CIRCAN_0 study. Oncotarget 2017, 8, 87980–87996. [Google Scholar] [CrossRef] [Green Version]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, Y.Y.; Chik, K.-W.; Chiu, R.W.; Ho, C.-Y.; Lam, C.W.; Lo, Y.D. Predominant Hematopoietic Origin of Cell-free DNA in Plasma and Serum after Sex-mismatched Bone Marrow Transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Hedtke, M.; Rejas, R.P.; Froelich, M.F.; Ast, V.; Duda, A.; Mirbach, L.; Costina, V.; Martens, U.M.; Hofheinz, R.D.; Neumaier, M.; et al. Liquid profiling of circulating tumor DNA in colorectal cancer: Steps needed to achieve its full clinical value as standard care. Mol. Oncol. 2021. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating Tumor DNA as a Liquid Biopsy for Cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef]
- Lui, Y.Y.; Dennis, Y.M. Circulating DNA in Plasma and Serum: Biology, Preanalytical Issues and Diagnostic Applications. Clin. Chem. Lab. Med. 2002, 40, 962–968. [Google Scholar] [CrossRef]
- Van Der Vaart, M.; Pretorius, P.J. Circulating DNA. Its origin and fluctuation. Ann. N. Y. Acad. Sci. 2008, 1137, 18–26. [Google Scholar] [CrossRef]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 827. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.-L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haselmann, V.; Gebhardt, C.; Brechtel, I.; Duda, A.; Czerwinski, C.; Sucker, A.; Holland-Letz, T.; Utikal, J.; Schadendorf, D.; Neumaier, M. Liquid Profiling of Circulating Tumor DNA in Plasma of Melanoma Patients for Companion Diagnostics and Monitoring of BRAF Inhibitor Therapy. Clin. Chem. 2018, 64, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Duan, J.; Cai, S.; Han, M.; Dong, H.; Zhao, J.; Zhu, B.; Wang, S.; Zhuo, M.; Sun, J.; et al. Assessment of Blood Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Patients With Non-Small Cell Lung Cancer With Use of a Next-Generation Sequencing Cancer Gene Panel. JAMA Oncol. 2019, 5, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Tzanikou, E.; Lianidou, E. The potential of ctDNA analysis in breast cancer. Crit. Rev. Clin. Lab. Sci. 2020, 57, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; García-Campelo, R.; de Álava, E.; Vera, R.; Rodríguez-Peralto, J.L.; Rodríguez-Lescure, Á.; Bellosillo, B.; Garrido, P.; Rojo, F.; Álvarez-Alegret, R. Liquid biopsy in oncology: A consensus statement of the Spanish Society of Pathology and the Spanish Society of Medical Oncology. Clin. Transl. Oncol. 2020, 22, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Siravegna, G.; Mussolin, B.; Venesio, T.; Marsoni, S.; Seoane, J.; Dive, C.; Papadopoulos, N.; Kopetz, S.; Corcoran, R.B.; Siu, L.L.; et al. How liquid biopsies can change clinical practice in oncology. Ann. Oncol. 2019, 30, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 2014, 20, 430–435. [Google Scholar] [CrossRef]
- Malapelle, U.; De-Las-Casas, C.M.; Rocco, D.; Garzon, M.; Pisapia, P.; Jordana-Ariza, N.; Russo, M.; Sgariglia, R.; De Luca, C.; Pepe, F.; et al. Development of a gene panel for next-generation sequencing of clinically relevant mutations in cell-free DNA from cancer patients. Br. J. Cancer 2017, 116, 802–810. [Google Scholar] [CrossRef] [Green Version]
- Qiu, M.; Wang, J.; Xu, Y.; Ding, X.; Li, M.; Jiang, F.; Xu, L.; Yin, R. Circulating Tumor DNA Is Effective for the Detection of EGFR Mutation in Non-Small Cell Lung Cancer: A Meta-analysis. Cancer Epidemiol. Biomark. Prev. 2015, 24, 206–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.-X.; Fu, Q.; Guo, Y.-Y.; Ye, M.; Zhao, H.-X.; Wang, Q.; Peng, X.-M.; Li, Q.-W.; Wang, R.-L.; Xiao, W.-H. Effectiveness of circulating tumor DNA for detection of KRAS gene mutations in colorectal cancer patients: A meta-analysis. OncoTargets Ther. 2017, 10, 945–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Xie, L.; Song, X. The diagnostic accuracy of circulating free DNA for the detection of KRAS mutation status in colorectal cancer: A meta-analysis. Cancer Med. 2019, 8, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Shen, L.; Zheng, D. Diagnostic value of circulating free DNA for the detection of EGFR mutation status in NSCLC: A systematic review and meta-analysis. Sci. Rep. 2014, 4, srep06269. [Google Scholar] [CrossRef]
- Tang, M.; Deng, Z.; Li, B.; Peng, Y.; Song, M.; Liu, J. Circulating Tumor DNA is Effective for Detection of KRAS Mutation in Colorectal Cancer: A Meta-Analysis. Int. J. Biol. Markers 2017, 32, e421–e427. [Google Scholar] [CrossRef]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef]
- Montagut, C.; Tsui, D.W.; Diaz, L.A., Jr. Detection of somatic RAS mutations in circulating tumor DNA from metastatic colorectal cancer patients: Are we ready for clinical use? Ann. Oncol. 2018, 29, 1083–1084. [Google Scholar] [CrossRef]
- Jenkins, S.; Yang, J.C.-H.; Ramalingam, S.S.; Yu, K.; Patel, S.; Weston, S.; Hodge, R.; Cantarini, M.; Jänne, P.A.; Mitsudomi, T.; et al. Plasma ctDNA Analysis for Detection of the EGFR T790M Mutation in Patients with Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Madison, R.; Schrock, A.B.; Castellanos, E.; Gregg, J.P.; Snider, J.; Ali, S.M.; Miller, V.A.; Singal, G.; Alexander, B.M.; Venstrom, J.M.; et al. Retrospective analysis of real-world data to determine clinical outcomes of patients with advanced non-small cell lung cancer following cell-free circulating tumor DNA genomic profiling. Lung Cancer 2020, 148, 69–78. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remon, J.; Caramella, C.; Jovelet, C.; Lacroix, L.; Lawson, A.; Smalley, S.; Howarth, K.; Gale, D.; Green, E.; Plagnol, V.; et al. Osimertinib benefit in EGFR-mutant NSCLC patients with T790M-mutation detected by circulating tumour DNA. Ann. Oncol. 2017, 28, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Serna-Blasco, R.; Alfaro, C.; Sánchez-Herrero, E.; Barquín, M.; Turpin, M.C.; Chico, S.; Sanz-Moreno, S.; Rodrigez-Festa, A.; Laza-Briviesca, R.; et al. ctDNA analysis reveals different molecular patterns upon disease progression in patients treated with osimertinib. Transl. Lung Cancer Res. 2020, 9, 532–540. [Google Scholar] [CrossRef]
- Sharma, G.G.; Mota, I.; Mologni, L.; Patrucco, E.; Gambacorti-Passerini, C.; Chiarle, R. Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes. Cancers 2018, 10, 62. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Solomon, B.J.; Besse, B.; Bauer, T.M.; Lin, C.-C.; Soo, R.A.; Riely, G.J.; Ou, S.-H.I.; Clancy, J.S.; Li, S.; et al. ALK Resistance Mutations and Efficacy of Lorlatinib in Advanced Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2019, 37, 1370–1379. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, B.; Hrebien, S.; Morden, J.P.; Beaney, M.; Fribbens, C.; Huang, X.; Liu, Y.; Bartlett, C.H.; Koehler, M.; Cristofanilli, M.; et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat. Commun. 2018, 9, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools (accessed on 8 February 2022).
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Song, Y.; Hu, C.; Xie, Z.; Wu, L.; Zhu, Z.; Rao, C.; Liu, L.; Chen, Y.; Liang, N.; Chen, J.; et al. Circulating tumor DNA clearance predicts prognosis across treatment regimen in a large real-world longitudinally monitored advanced non-small cell lung cancer cohort. Transl. Lung Cancer Res. 2020, 9, 269–279. [Google Scholar] [CrossRef]
- Andersson, D.; Kristiansson, H.; Kubista, M.; Ståhlberg, A. Ultrasensitive circulating tumor DNA analysis enables precision medicine: Experimental workflow considerations. Expert Rev. Mol. Diagn. 2021, 21, 299–310. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Cohen, J.D.; Kinde, I.; Ptak, J.; Popoli, M.; Schaefer, J.; Silliman, N.; Dobbyn, L.; Tie, J.; et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncol. 2019, 5, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.K.; Wu, H.-T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parseghian, C.M.; Loree, J.M.; Morris, V.K.; Liu, X.; Clifton, K.K.; Napolitano, S.; Henry, J.T.; Pereira, A.A.; Vilar, E.; Johnson, B.; et al. Anti-EGFR-resistant clones decay exponentially after progression: Implications for anti-EGFR re-challenge. Ann. Oncol. 2019, 30, 243–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, S.B.; Narayan, A.; Kole, A.J.; Decker, R.H.; Teysir, J.; Carriero, N.J.; Lee, A.; Nemati, R.; Nath, S.K.; Mane, S.M.; et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin. Cancer Res. 2018, 24, 1872–1880. [Google Scholar] [CrossRef] [Green Version]
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Stehr, H.; Craig, A.F.M.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med. 2016, 8, 364ra155. [Google Scholar] [CrossRef] [Green Version]
- Reinert, T.; Schøler, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.-S.; Mortensen, F.V.; Stribolt, K.; et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2016, 65, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef]
- Basnet, S.; Zhang, Z.-Y.; Liao, W.-Q.; Li, S.-H.; Li, P.-S.; Ge, H.-Y. The Prognostic Value of Circulating Cell-Free DNA in Colorectal Cancer: A Meta-Analysis. J. Cancer 2016, 7, 1105–1113. [Google Scholar] [CrossRef] [Green Version]
- Spindler, K.-L.G.; Boysen, A.K.; Pallisgård, N.; Johansen, J.S.; Tabernero, J.; Sørensen, M.M.; Jensen, B.V.; Hansen, T.F.; Sefrioui, D.; Andersen, R.F.; et al. Cell-Free DNA in Metastatic Colorectal Cancer: A Systematic Review and Meta-Analysis. Oncologist 2017, 22, 1049–1055. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Peng, J.; Xiao, Q.; Wu, H.-X.; Wu, X.; Wang, F.; Li, L.; Ding, P.; Zhao, Q.; Li, Y.; et al. Postoperative circulating tumor DNA as markers of recurrence risk in stages II to III colorectal cancer. J. Hematol. Oncol. 2021, 14, 80. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Nakamura, Y.; Kotani, D.; Yukami, H.; Mishima, S.; Sawada, K.; Shirasu, H.; Ebi, H.; Yamanaka, T.; Aleshin, A.; et al. CIRCULATE-Japan: Circulating tumor DNA-guided adaptive platform trials to refine adjuvant therapy for colorectal cancer. Cancer Sci. 2021, 112, 2915–2920. [Google Scholar] [CrossRef]
- Olsson, E.; Winter, C.; George, A.; Chen, Y.; Howlin, J.; Tang, M.-H.E.; Dahlgren, M.; Schulz, R.; Grabau, D.; van Westen, D.; et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 2015, 7, 1034–1047. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, L.; Douville, C.; Cohen, J.D.; Yen, T.-T.; Kinde, I.; Sundfelt, K.; Kjær, S.K.; Hruban, R.H.; Shih, I.-M.; et al. Evaluation of liquid from the Papanicolaou test and other liquid biopsies for the detection of endometrial and ovarian cancers. Sci. Transl. Med. 2018, 10, eaap8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, Y.N.; Dhillon, S. Epi proColon((R)) 2.0 CE: A Blood-Based Screening Test for Colorectal Cancer. Mol. Diagn. Ther. 2017, 21, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Soria-Comes, T.; Palomar-Abril, V.; Ureste, M.M.; Guerola, M.T.; Maiques, I.C.M. Real-World Data of the Correlation between EGFR Determination by Liquid Biopsy in Non-squamous Non-small Cell Lung Cancer (NSCLC) and the EGFR Profile in Tumor Biopsy. Pathol. Oncol. Res. POR 2020, 26, 845–851. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Aucamp, J.; Pretorius, P.J. Cell-free DNA: Preanalytical variables. Clin. Chim. Acta 2015, 450, 243–253. [Google Scholar] [CrossRef]
- Meddeb, R.; Pisareva, E.; Thierry, A.R. Guidelines for the Preanalytical Conditions for Analyzing Circulating Cell-Free DNA. Clin. Chem. 2019, 65, 623–633. [Google Scholar] [CrossRef]
- Ijzerman, M.; de Boer, J.; Azad, A.; Degeling, K.; Geoghegan, J.; Hewitt, C.; Hollande, F.; Lee, B.; To, Y.H.; Tothill, R.W.; et al. Towards Routine Implementation of Liquid Biopsies in Cancer Management: It Is Always Too Early, until Suddenly It Is Too Late. Diagnostics 2021, 11, 103. [Google Scholar] [CrossRef]
- Salk, J.J.; Loubet-Senear, K.; Maritschnegg, E.; Valentine, C.C.; Williams, L.N.; Higgins, J.E.; Horvat, R.; Vanderstichele, A.; Nachmanson, D.; Baker, K.T.; et al. Ultra-Sensitive TP53 Sequencing for Cancer Detection Reveals Progressive Clonal Selection in Normal Tissue over a Century of Human Lifespan. Cell Rep. 2019, 28, 132–144.e133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.C.; Sale, J.E.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular In Vitro Diagnostic Examinations—Specifications for Pre-Examination Processes for Venous Whole Blood—Part 3: Isolated Circulating Cell Free DNA from Plasma. CEN/TS 16835-3:2015.
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef]
- Lampignano, R.; Neumann, M.H.D.; Weber, S.; Kloten, V.; Herdean, A.; Voss, T.; Groelz, D.; Babayan, A.; Tibbesma, M.; Schlumpberger, M.; et al. Multicenter Evaluation of Circulating Cell-Free DNA Extraction and Downstream Analyses for the Development of Standardized (Pre)analytical Work Flows. Clin. Chem. 2020, 66, 149–160. [Google Scholar] [CrossRef]
- Chan, K.C.; Yeung, S.-W.; Lui, W.-B.; Rainer, T.H.; Lo, Y.M. Effects of Preanalytical Factors on the Molecular Size of Cell-Free DNA in Blood. Clin. Chem. 2005, 51, 781–784. [Google Scholar] [CrossRef]
- Lam, N.Y.; Rainer, T.H.; Chiu, R.W.; Lo, Y.M. EDTA Is a Better Anticoagulant than Heparin or Citrate for Delayed Blood Processing for Plasma DNA Analysis. Clin. Chem. 2004, 50, 256–257. [Google Scholar] [CrossRef]
- Nikolaev, S.; Lemmens, L.; Koessler, T.; Blouin, J.-L.; Nouspikel, T. Circulating tumoral DNA: Preanalytical validation and quality control in a diagnostic laboratory. Anal. Biochem. 2018, 542, 34–39. [Google Scholar] [CrossRef]
- Risberg, B.; Tsui, D.W.Y.; Biggs, H.; Ruiz-Valdepenas Martin de Almagro, A.; Dawson, S.-J.; Hodgkin, C.; Jones, L.; Parkinson, C.; Piskorz, A.; Marass, F.; et al. Effects of Collection and Processing Procedures on Plasma Circulating Cell-Free DNA from Cancer Patients. J. Mol. Diagn. 2018, 20, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Kang, Q.; Henry, N.L.; Paoletti, C.; Jiang, H.; Vats, P.; Chinnaiyan, A.M.; Hayes, D.F.; Merajver, S.D.; Rae, J.M.; Tewari, M. Comparative analysis of circulating tumor DNA stability In K3EDTA, Streck, and CellSave blood collection tubes. Clin. Biochem. 2016, 49, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Parpart-Li, S.; Bartlett, B.; Popoli, M.; Adleff, V.; Tucker, L.; Steinberg, R.; Georgiadis, A.; Phallen, J.; Brahmer, J.R.; Azad, N.; et al. The Effect of Preservative and Temperature on the Analysis of Circulating Tumor DNA. Clin. Cancer Res. 2017, 23, 2471–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, R.W.K.; Poon, L.L.; Lau, T.K.; Leung, T.N.; Wong, E.M.C.; Lo, Y.M.D. Effects of Blood-Processing Protocols on Fetal and Total DNA Quantification in Maternal Plasma. Clin. Chem. 2001, 47, 1607–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallone, L.; Aldamry, M.; LaFleur, J.; Lan, C.; Ginestet, P.G.; Alirezaie, N.; Ferrario, C.; Aguilar-Mahecha, A.; Basik, M. A Study of Pre-Analytical Variables and Optimization of Extraction Method for Circulating Tumor DNA Measurements by Digital Droplet PCR. Cancer Epidemiol. Biomark. Prev. 2019, 28, 909–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, K.; Guttery, D.S.; Zahra, N.; Primrose, L.; Elshaw, S.R.; Pringle, J.H.; Blighe, K.; Marchese, S.D.; Hills, A.; Woodley, L.; et al. Influence of Plasma Processing on Recovery and Analysis of Circulating Nucleic Acids. PLoS ONE 2013, 8, e77963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ginkel, J.H.; van den Broek, D.A.; Van Kuik, J.; Linders, D.; De Weger, R.; Willems, S.M.; Huibers, M.M.H. Preanalytical blood sample workup for cell-free DNA analysis using Droplet Digital PCR for future molecular cancer diagnostics. Cancer Med. 2017, 6, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- El Messaoudi, S.; Rolet, F.; Mouliere, F.; Thierry, A.R. Circulating cell free DNA: Preanalytical considerations. Clin. Chim. Acta 2013, 424, 222–230. [Google Scholar] [CrossRef]
- Devonshire, A.S.; Whale, A.S.; Gutteridge, A.; Jones, G.; Cowen, S.; Foy, C.A.; Huggett, J.F. Towards standardisation of cell-free DNA measurement in plasma: Controls for extraction efficiency, fragment size bias and quantification. Anal. Bioanal. Chem. 2014, 406, 6499–6512. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, R.J.; Lee, J.H.; Kefford, R.F.; Rizos, H. Evaluation of commercial kits for purification of circulating free DNA. Cancer Genet. 2018, 228-229, 21–27. [Google Scholar] [CrossRef]
- Van Der Leest, P.; Schuuring, E. The potential of combined mutation sequencing of plasma circulating cell-free DNA and matched white blood cells for treatment response prediction. Mol. Oncol. 2020, 14, 487–489. [Google Scholar] [CrossRef]
- Xue, X.; Teare, M.D.; Holen, I.; Zhu, Y.M.; Woll, P.J. Optimizing the yield and utility of circulating cell-free DNA from plasma and serum. Clin. Chim. Acta 2009, 404, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorber, L.; Zwaenepoel, K.; Deschoolmeester, V.; Roeyen, G.; Lardon, F.; Rolfo, C.; Pauwels, P. A Comparison of Cell-Free DNA Isolation Kits: Isolation and Quantification of Cell-Free DNA in Plasma. J. Mol. Diagn. 2017, 19, 162–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haselmann, V.; Ahmad-Nejad, P.; Geilenkeuser, W.J.; Duda, A.; Gabor, M.; Eichner, R.; Patton, S.; Neumaier, M. Results of the first external quality assessment scheme (EQA) for isolation and analysis of circulating tumour DNA (ctDNA). Clin. Chem. Lab. Med. 2018, 56, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Bartels, S.; Persing, S.; Hasemeier, B.; Schipper, E.; Kreipe, H.; Lehmann, U. Molecular Analysis of Circulating Cell-Free DNA from Lung Cancer Patients in Routine Laboratory Practice: A Cross-Platform Comparison of Three Different Molecular Methods for Mutation Detection. J. Mol. Diagn. 2017, 19, 722–732. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [Green Version]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef]
- Haselmann, V.; Geilenkeuser, W.J.; Helfert, S.; Eichner, R.; Hetjens, S.; Neumaier, M.; Ahmad-Nejad, P. Thirteen Years of an International External Quality Assessment Scheme for Genotyping: Results and Recommendations. Clin. Chem. 2016, 62, 1084–1095. [Google Scholar] [CrossRef] [Green Version]
- Weber, S.; Spiegl, B.; Perakis, S.O.; Ulz, C.M.; Abuja, P.M.; Kashofer, K.; Van Der Leest, P.; Azpurua, M.A.; Tamminga, M.; Brudzewsky, D.; et al. Technical Evaluation of Commercial Mutation Analysis Platforms and Reference Materials for Liquid Biopsy Profiling. Cancers 2020, 12, 1588. [Google Scholar] [CrossRef]
- Pös, Z.; Pös, O.; Styk, J.; Mocova, A.; Strieskova, L.; Budis, J.; Kadasi, L.; Radvanszky, J.; Szemes, T. Technical and Methodological Aspects of Cell-Free Nucleic Acids Analyzes. Int. J. Mol. Sci. 2020, 21, 8634. [Google Scholar] [CrossRef]
- Godsey, J.H.; Silvestro, A.; Barrett, J.C.; Bramlett, K.; Chudova, D.; Deras, I.; Dickey, J.; Hicks, J.; Johann, D.J.; Leary, R.; et al. Generic Protocols for the Analytical Validation of Next-Generation Sequencing-Based ctDNA Assays: A Joint Consensus Recommendation of the BloodPAC’s Analytical Variables Working Group. Clin. Chem. 2020, 66, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.X.; He, Y.; Sanford, E.; Montesion, M.; Frampton, G.M.; Vignot, S.; Soria, J.-C.; Ross, J.S.; Miller, V.A.; Stephens, P.J.; et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Comput. Biol. 2018, 14, e1005965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, A.; Morris, V.K.; Allegra, C.J.; Atreya, C.; Benson, A.B., 3rd; Boland, P.; Chung, K.; Copur, M.S.; Corcoran, R.B.; Deming, D.A.; et al. ctDNA applications and integration in colorectal cancer: An NCI Colon and Rectal–Anal Task Forces whitepaper. Nat. Rev. Clin. Oncol. 2020, 17, 757–770. [Google Scholar] [CrossRef]
- Miller, W.G.; Jones, G.R.; Horowitz, G.L.; Weykamp, C. Proficiency Testing/External Quality Assessment: Current Challenges and Future Directions. Clin. Chem. 2011, 57, 1670–1680. [Google Scholar] [CrossRef] [Green Version]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar] [PubMed]
- Streubel, A.; Stenzinger, A.; Stephan-Falkenau, S.; Kollmeier, J.; Misch, D.; Blum, T.G.; Bauer, T.; Landt, O.; Ende, A.A.; Schirmacher, P.; et al. Comparison of different semi-automated cfDNA extraction methods in combination with UMI-based targeted sequencing. Oncotarget 2019, 10, 5690–5702. [Google Scholar] [CrossRef] [PubMed]
- Markus, H.; Contente-Cuomo, T.; Farooq, M.; Liang, W.S.; Borad, M.J.; Sivakumar, S.; Gollins, S.; Tran, N.L.; Dhruv, H.D.; Berens, M.E.; et al. Evaluation of pre-analytical factors affecting plasma DNA analysis. Sci. Rep. 2018, 8, 7375. [Google Scholar] [CrossRef] [Green Version]
- Bernabé, R.; Hickson, N.; Wallace, A.; Blackhall, F.H. What do we need to make circulating tumour DNA (ctDNA) a routine diagnostic test in lung cancer? Eur. J. Cancer 2017, 81, 66–73. [Google Scholar] [CrossRef]
- Douglas, M.P.; Gray, S.W.; Phillips, K.A. Private Payer and Medicare Coverage for Circulating Tumor DNA Testing: A Historical Analysis of Coverage Policies From 2015 to 2019. J. Natl. Compr. Cancer Netw. 2020, 18, 866–872. [Google Scholar] [CrossRef]


{kind=link}
| Gene | Alteration | Cancer Type |
|---|---|---|
| ALK | Fusions, Oncogenic Mutations | NSCLC |
| ATM | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| BARD1 | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| BRAF | V600 | Melanoma |
| BRAF | V600E | Anaplastic Thyroid Cancer, CRC, NSCLC |
| BRAF | V600E, V600K | Melanoma |
| BRCA1 | Oncogenic Mutations | Ovary/Fallopian Tube, Ovarian Cancer, Peritoneal Serous Carcinoma, Prostate Cancer, NOS, Prostate Cancer |
| BRCA2 | Oncogenic Mutations | Ovary/Fallopian Tube, Ovarian Cancer, Peritoneal Serous Carcinoma, Prostate Cancer, NOS, Prostate Cancer |
| BRIP1 | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| CDK12 | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| CHEK1 | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| CHEK2 | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| EGFR | Exon 19 deletion, L858R | NSCLC |
| EGFR | Exon 20 insertion | NSCLC |
| EGFR | G719 | NSCLC |
| EGFR | L861Q | NSCLC |
| EGFR | S768I | NSCLC |
| EGFR | T790M | NSCLC |
| ERBB2 | Amplification | Breast Cancer, Esophagogastric Cancer |
| FANCL | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| FGFR2 | Fusions | Bladder Cancer, Cholangiocarcinoma |
| FGFR3 | Fusions | Bladder Cancer |
| FGFR3 | G370C, R248C, S249C, Y373C | Bladder Cancer |
| IDH1 | R132 | Cholangiocarcinoma, Intrahepatic Cholangiocarcinoma |
| KIT | A502,Y503dup, K509I, N505I, S476I, S501, A502dup | Gastrointestinal Stromal Tumor |
| KIT | A829P and 5 other alterations | Gastrointestinal Stromal Tumor |
| KIT | D572A and 65 other alterations | Gastrointestinal Stromal Tumor |
| KIT | K642E | Gastrointestinal Stromal Tumor |
| KIT | T670I | Gastrointestinal Stromal Tumor |
| KIT | V654A | Gastrointestinal Stromal Tumor |
| KRAS | G12C | NSCLC |
| KRAS | Wildtype | CRC |
| MET | D1010, Exon 14 deletion, Exon 14 splice mutation | NSCLC |
| NF1 | Oncogenic Mutations | Neurofibroma |
| NRAS | Wildtype | CRC |
| NTRK1 | Fusions | All Solid Tumors |
| NTRK2 | Fusions | All Solid Tumors |
| NTRK3 | Fusions | All Solid Tumors |
| PALB2 | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| PDGFB | COL1A1-PDGFB Fusion | Dermatofibrosarcoma Protuberans |
| PDGFRA | Exon 18 in-frame deletions, Exon 18 in-frame insertions, Exon 18 missense mutations | Gastrointestinal Stromal Tumor |
| PIK3CA | C420R and 10 other alterations | Breast Cancer |
| RAD51B | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| RAD51C | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| RAD51D | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| RAD54L | Oncogenic Mutations | Prostate Cancer, NOS, Prostate Cancer |
| RET | Fusions | NSCLC, Thyroid Cancer |
| RET | Oncogenic Mutations | Medullary Thyroid Cancer |
| ROS1 | Fusions | Non-Small Cell Lung Cancer |
| SMARCB1 | Deletion | Epithelioid Sarcoma |
| Company | Test | Method | Indication |
|---|---|---|---|
| Roche | cobas EGFR Mutation test v2 | qPCR | Detection of EGFR driver mutations in patients who may benefit from tyrosine kinase inhibitor (TKI) treatment |
| Qiagen | therascreen PIK3CA RGQ PCR Kit | qPCR | PIK3CA mutation detection in liquid biopsy for postmenopausal, hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2-)-negative advanced breast cancer patients |
| Guardant Health | Guradant360 CDx | NGS | Detection of EGFR and KRAS mutations eligible for FDA-approved treatment in patients with NSCLC |
| Foundation Medicine | FoundationOne Liquid CDx | NGS | Used as a companion diagnostic to identify patients (with NSCLC, prostate cancer, ovarian cancer, breast cancer) who may benefit from treatment with targeted therapies |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haselmann, V.; Hedtke, M.; Neumaier, M. Liquid Profiling for Cancer Patient Stratification in Precision Medicine—Current Status and Challenges for Successful Implementation in Standard Care. Diagnostics 2022, 12, 748. https://doi.org/10.3390/diagnostics12030748
Haselmann V, Hedtke M, Neumaier M. Liquid Profiling for Cancer Patient Stratification in Precision Medicine—Current Status and Challenges for Successful Implementation in Standard Care. Diagnostics. 2022; 12(3):748. https://doi.org/10.3390/diagnostics12030748
Chicago/Turabian StyleHaselmann, Verena, Maren Hedtke, and Michael Neumaier. 2022. "Liquid Profiling for Cancer Patient Stratification in Precision Medicine—Current Status and Challenges for Successful Implementation in Standard Care" Diagnostics 12, no. 3: 748. https://doi.org/10.3390/diagnostics12030748