Somatostatinoma and Neurofibromatosis Type 1-A Case Report and Review of the Literature



{kind=link}
{kind=link}
{kind=link}
{kind=link}
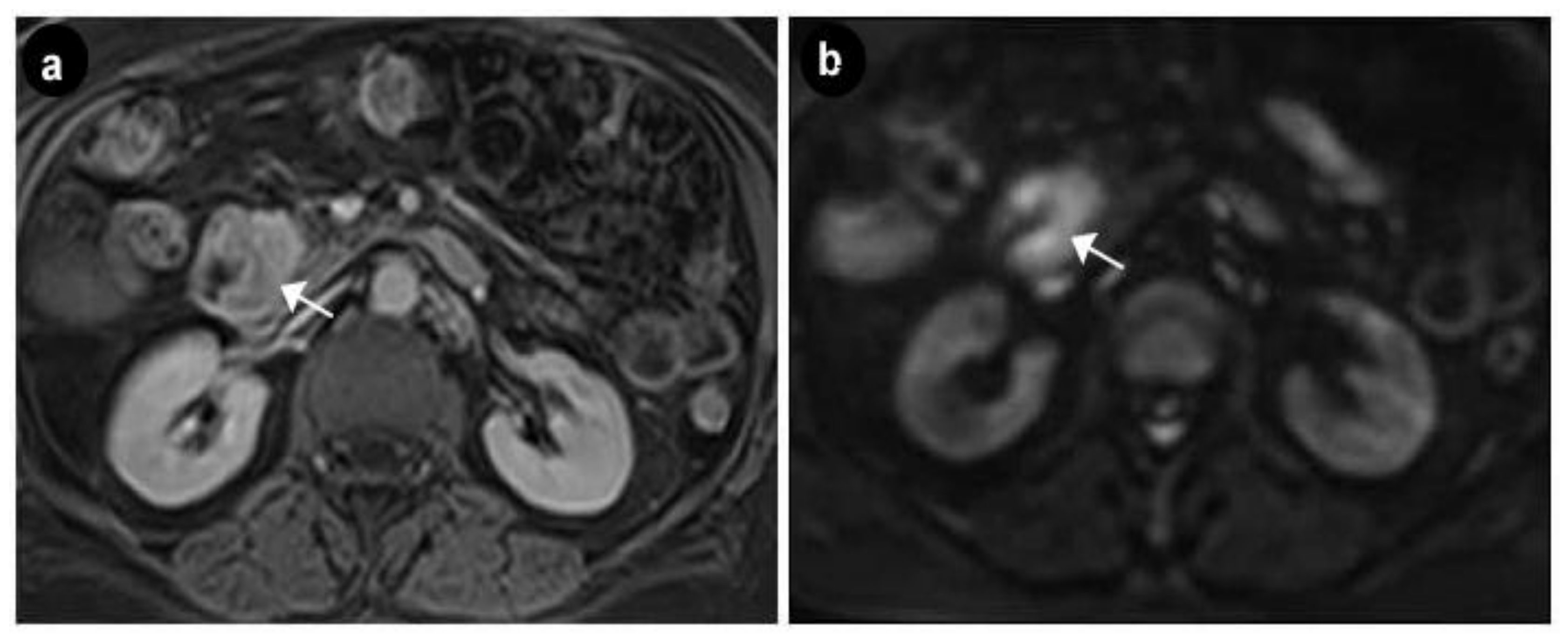{kind=link}
{kind=link}
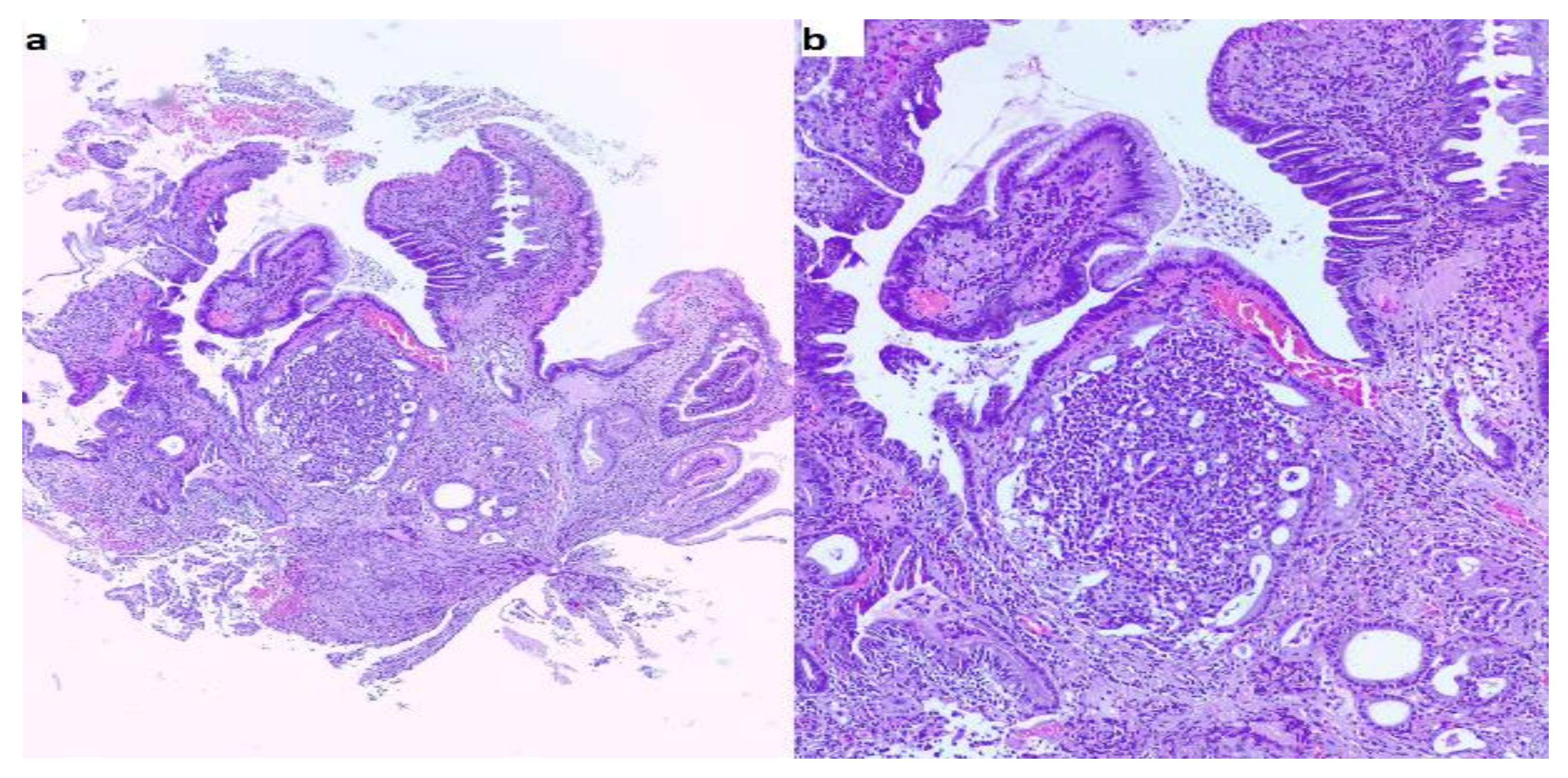{kind=link}
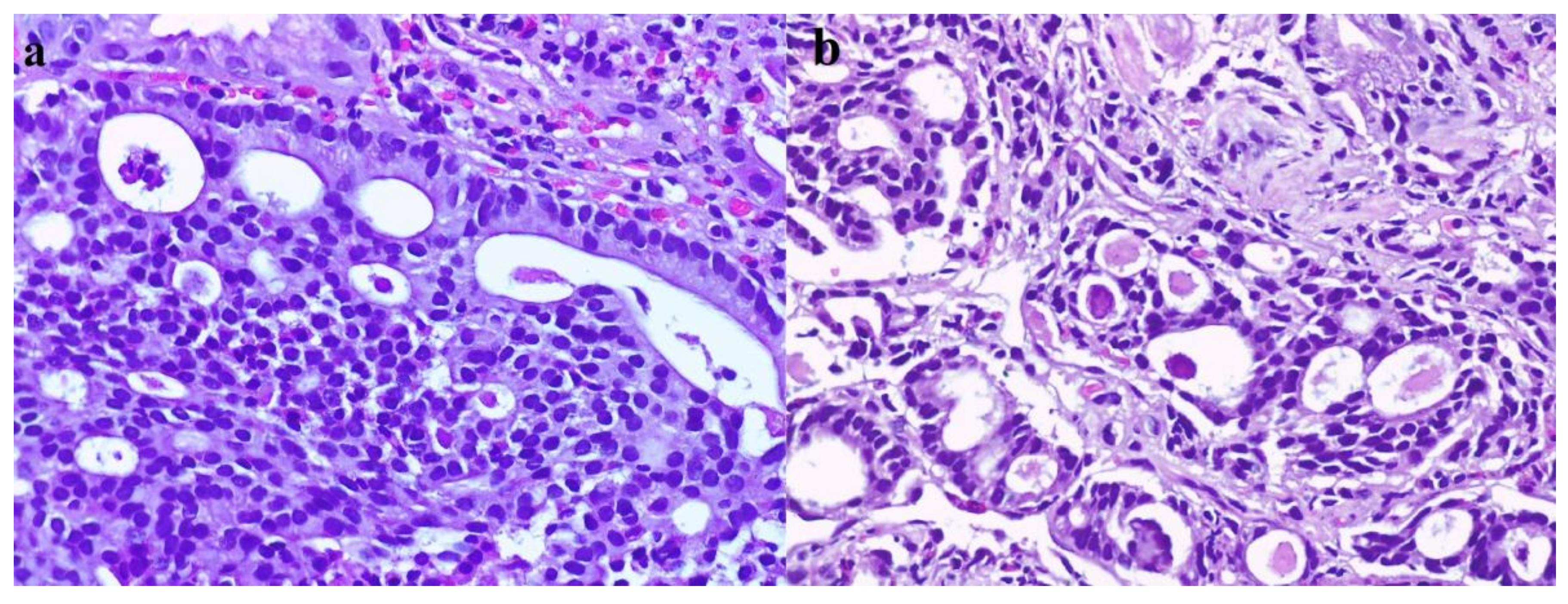{kind=link}
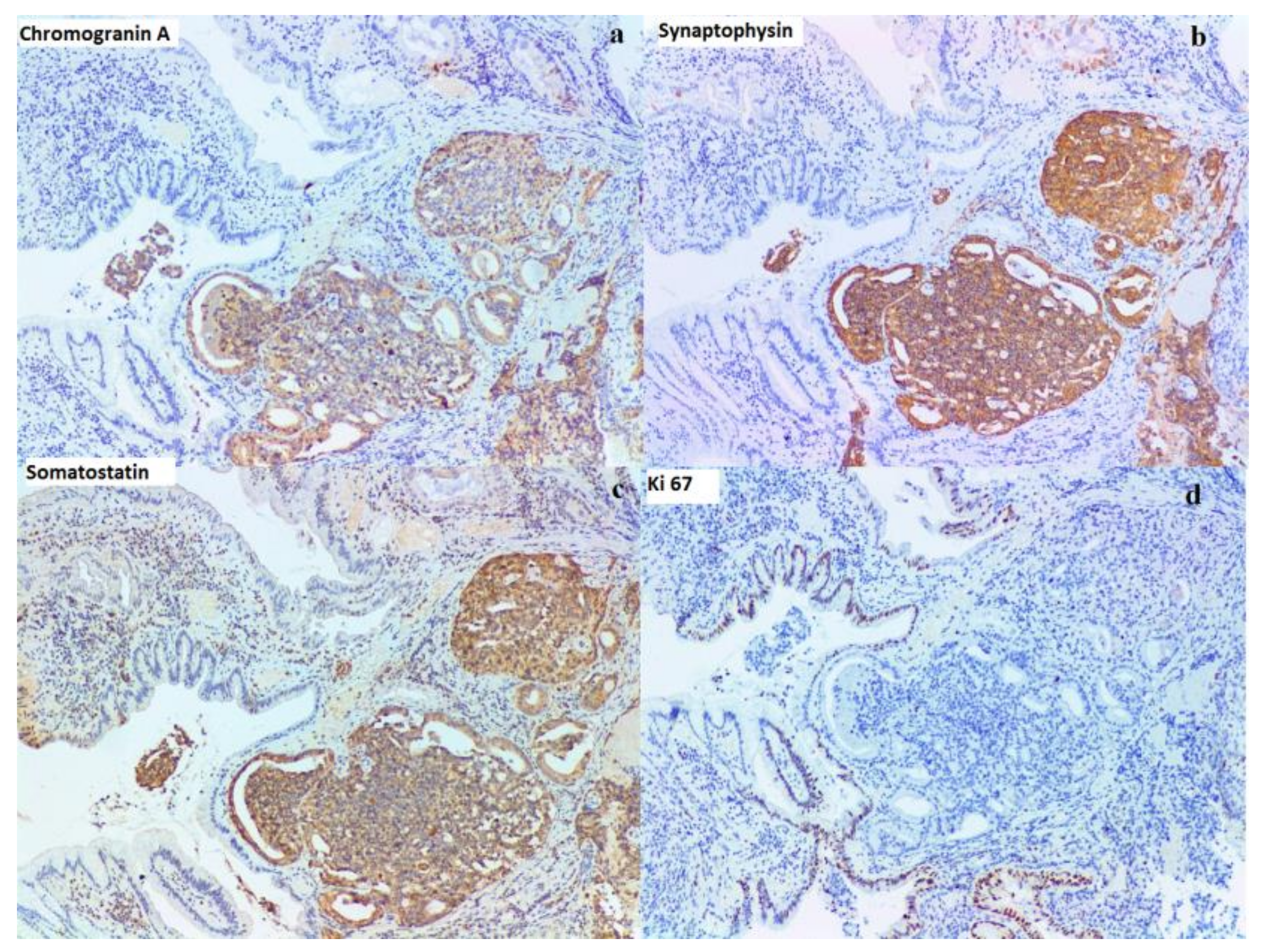{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
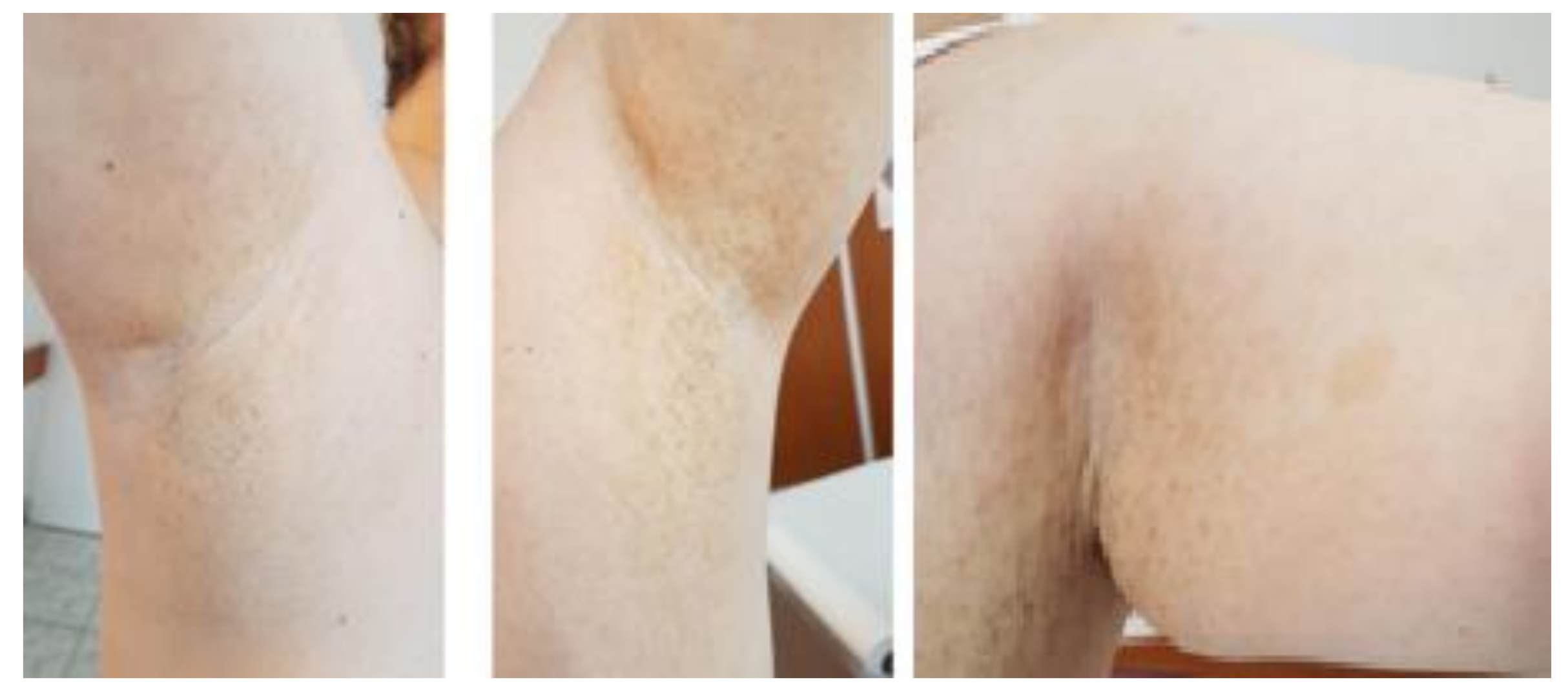
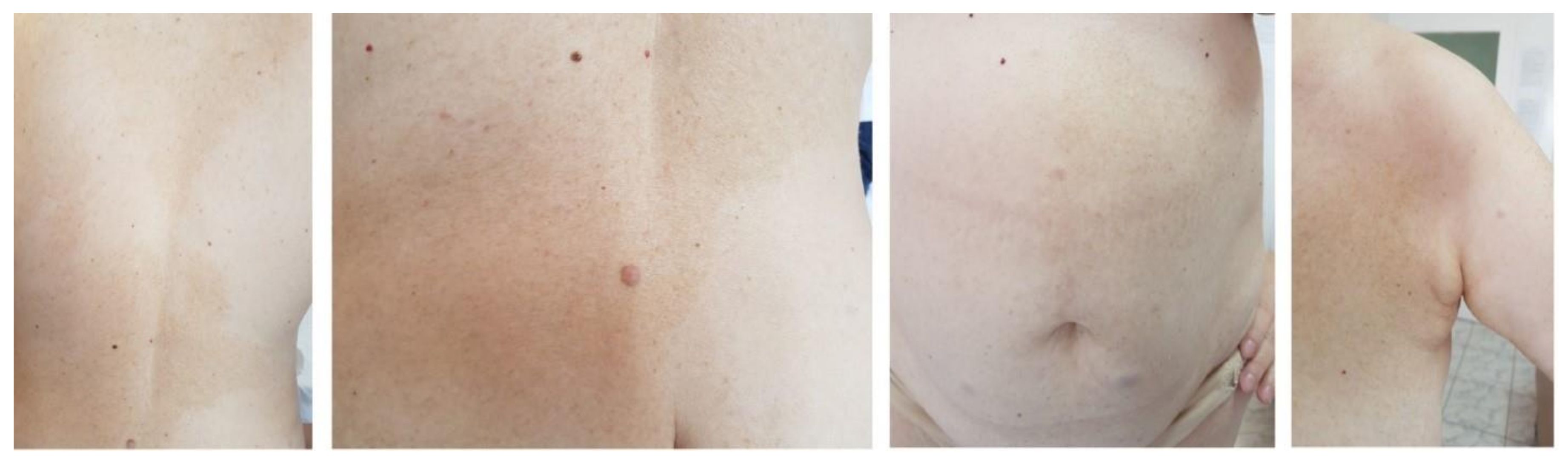
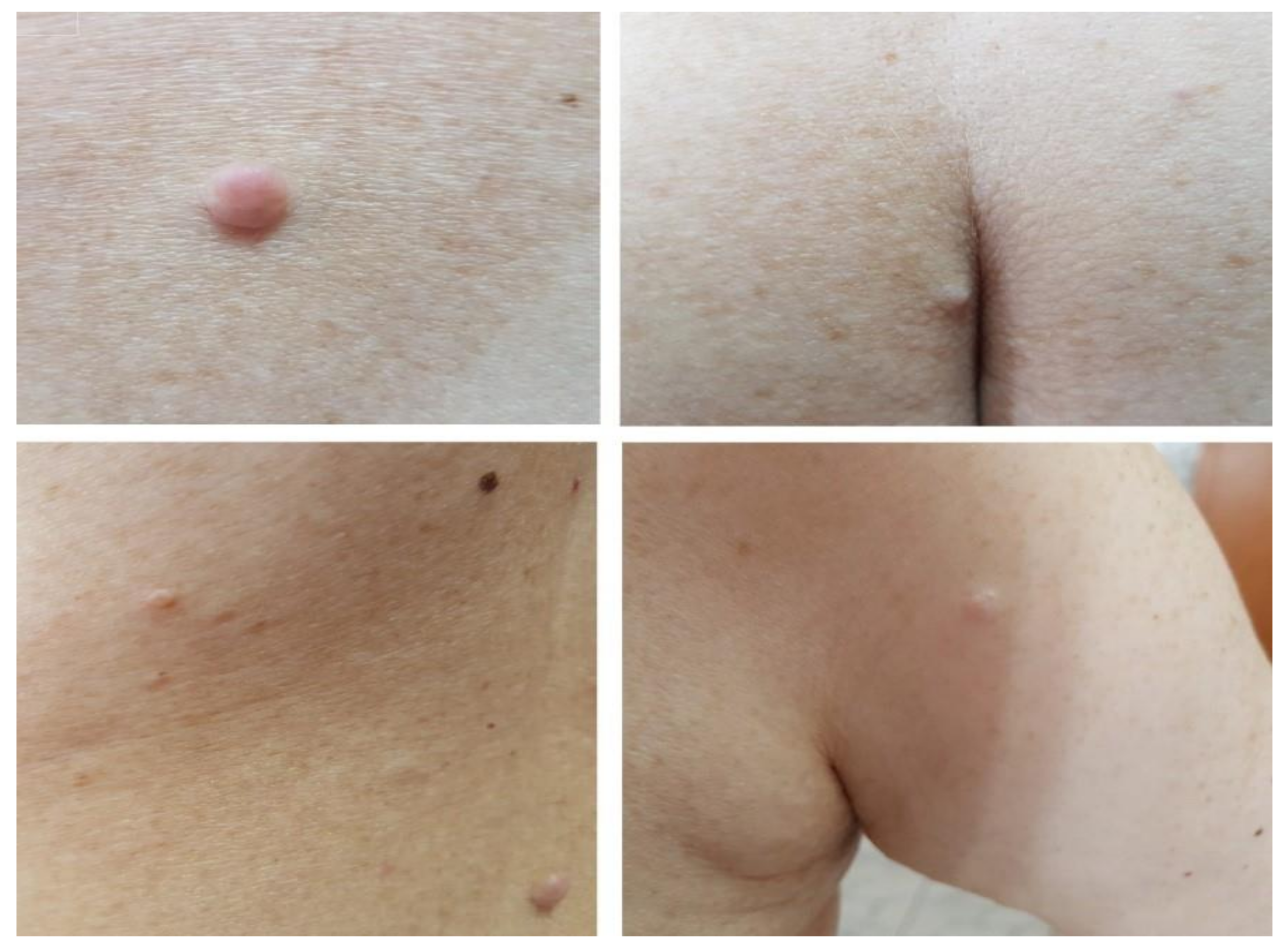
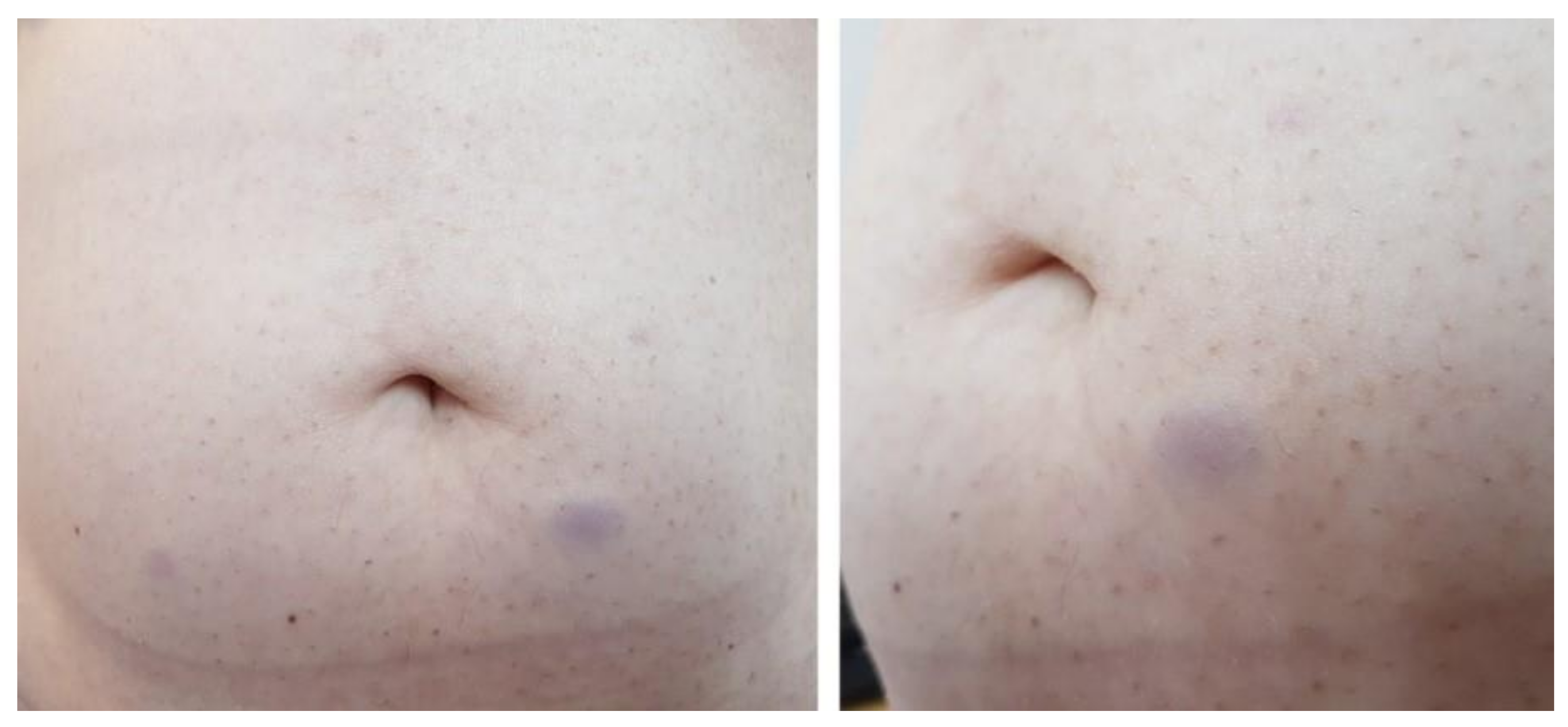
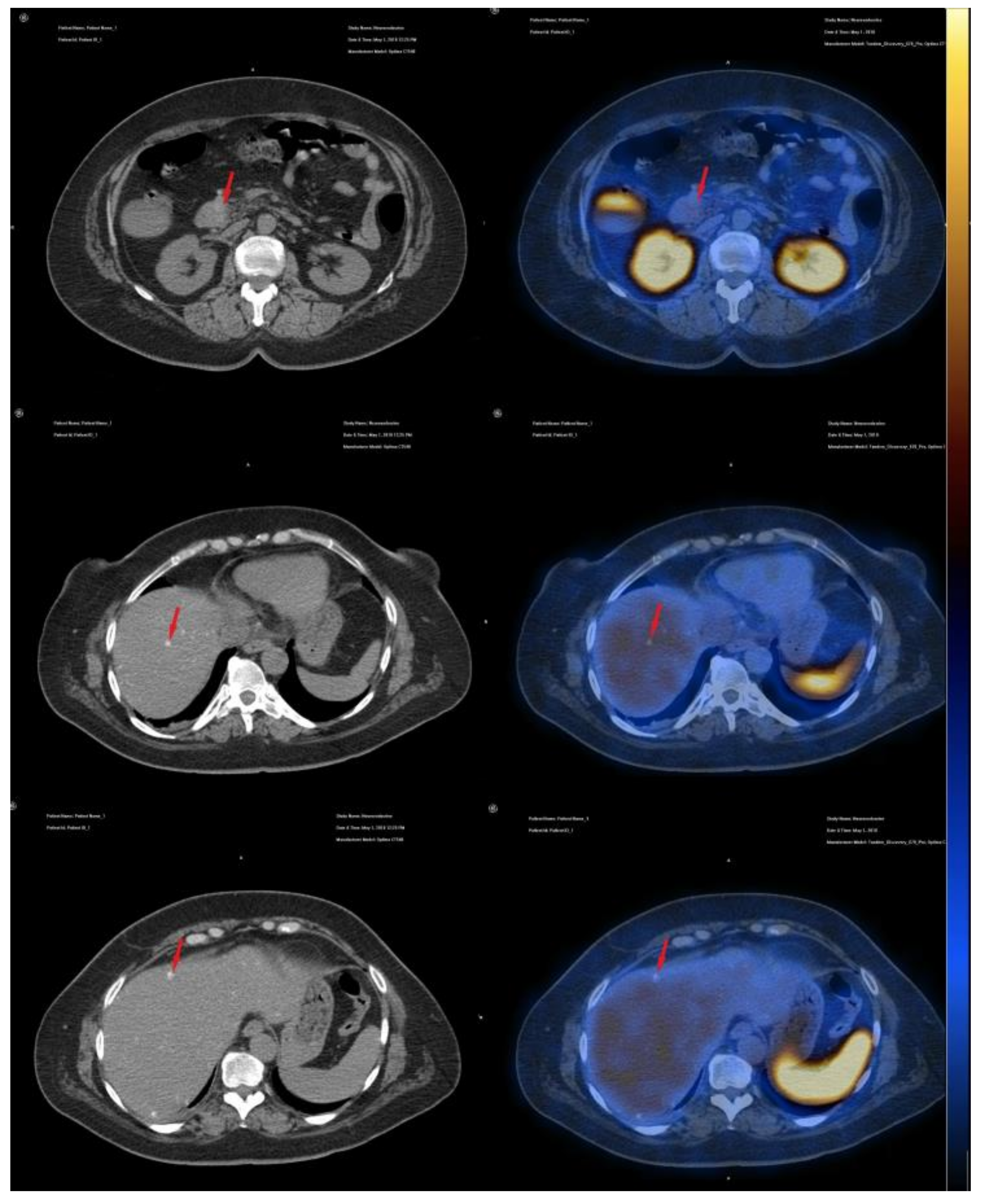
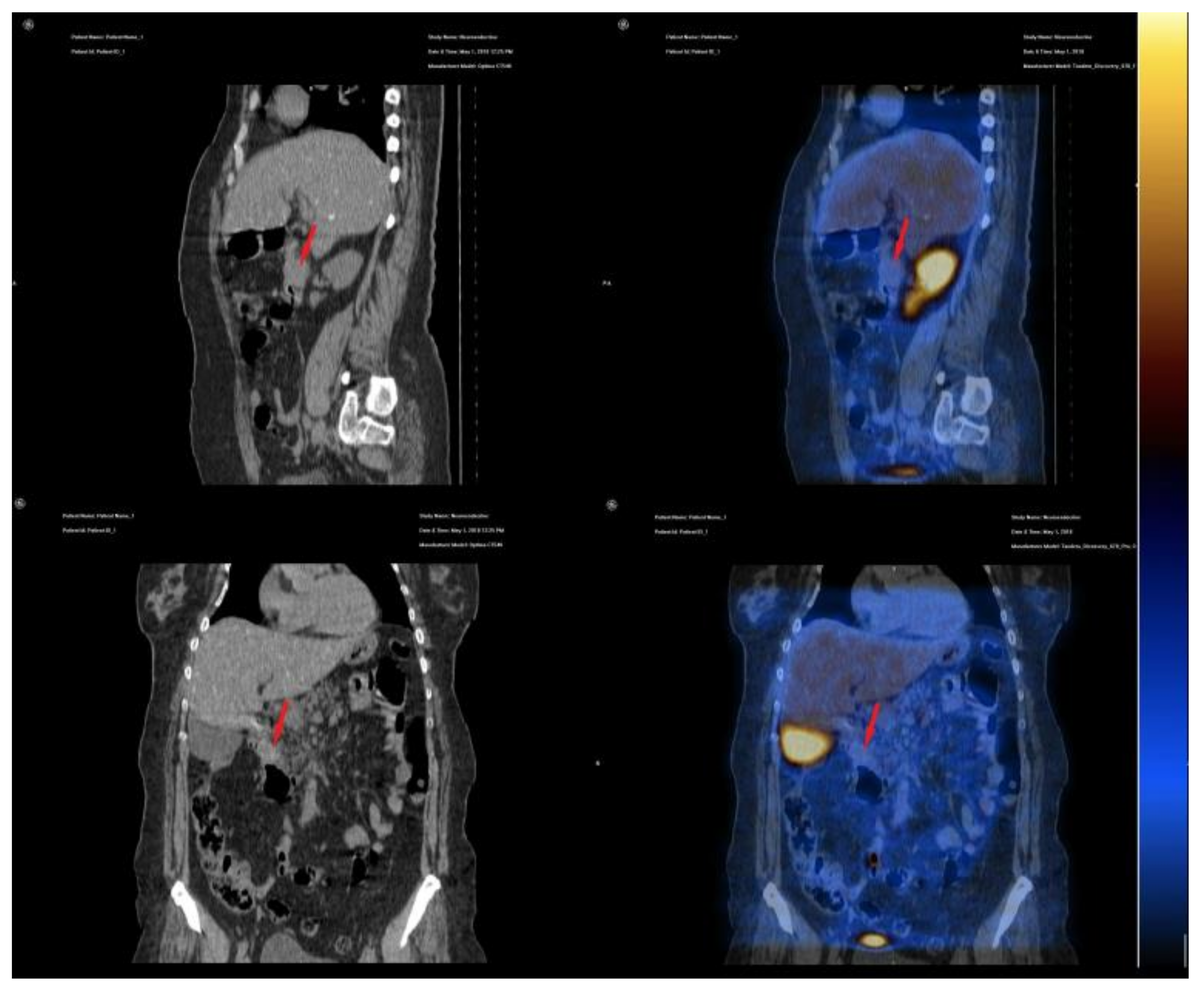
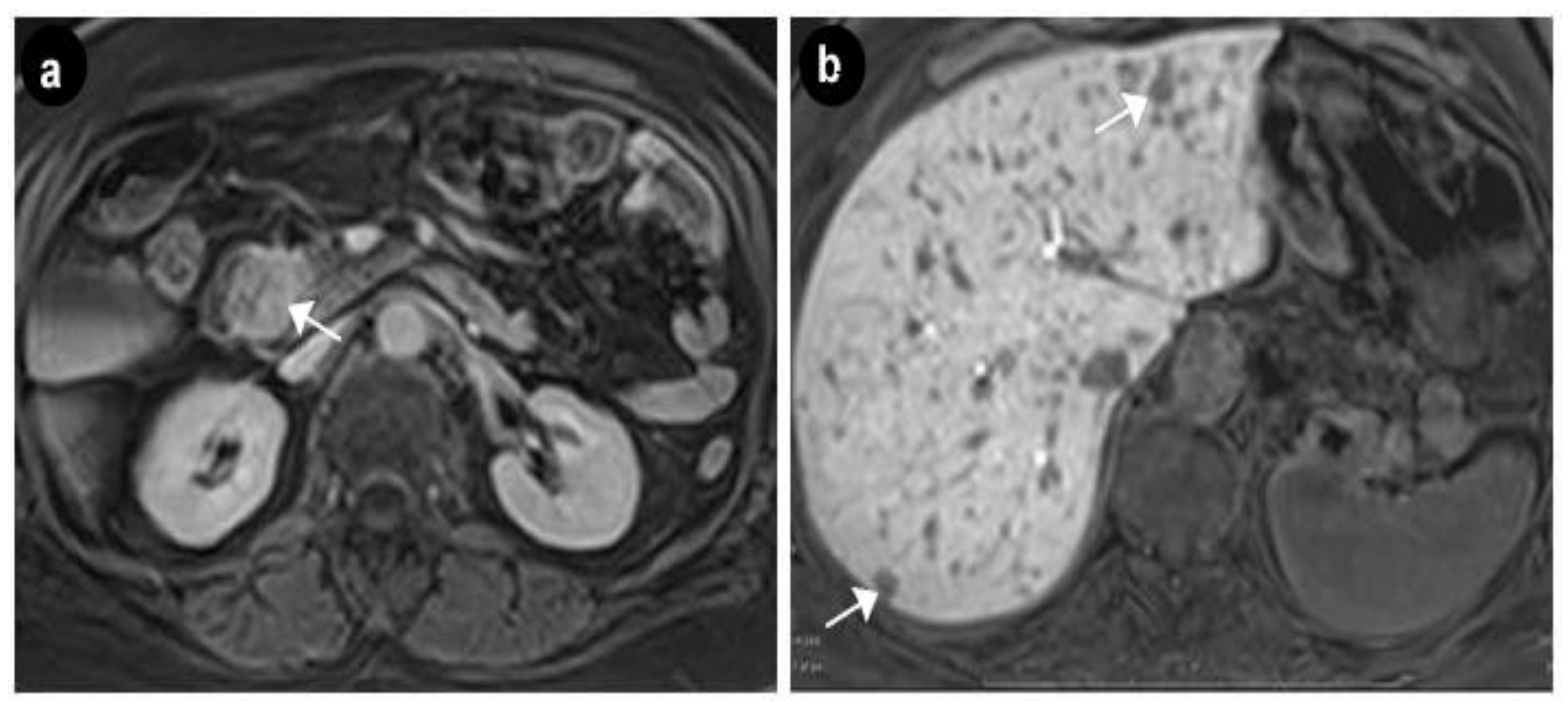
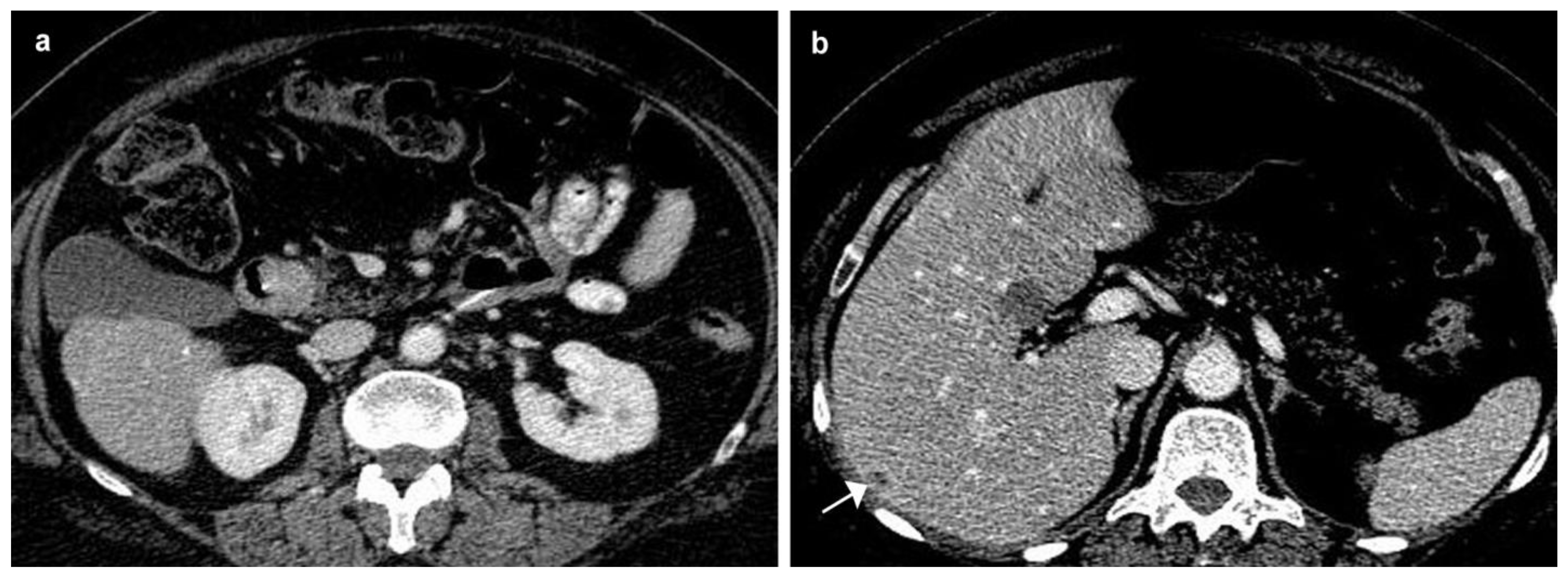
2. Case Presentation
3. Discussion
4. Conclusions
Funding
Conflicts of Interest
References
- Friesen, S.R. Update on the diagnosis and treatment of rare neuroendocrine tumors. Surg. Clin. North. Am. 1987, 67, 379–393. [Google Scholar] [CrossRef]
- Harris, G.J.; Tio, F.; Cruz, A.B., Jr. Somatostatinoma: A case report and review of the literature. J. Surg. Oncol. 1987, 36, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Klöppel, G.; Anlauf, M. Epidemiology, tumour biology and histopathological classification of neuroendocrine tumours of the gastrointestinal tract. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Soga, J.; Yakuwa, Y. Somatostatinoma/inhibitory syndrome: A statistical evaluation of 173 reported cases as compared to other pancreatic endocrinomas. J. Exp. Clin. Cancer Res. 1999, 18, 13–22. [Google Scholar]
- Doherty, G.M. Rare endocrine tumours of the GI tract. Best practice & research. Clin. Gastroenterol. 2005, 19, 807–817. [Google Scholar] [CrossRef]
- Vinik, A.; Pacak, K.; Feliberti, E.; Perry, R.R. Somatostatinoma. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2017. [Google Scholar]
- Agaimy, A.; Vassos, N.; Croner, R.S. Gastrointestinal manifestations of neurofibromatosis type 1 (Recklinghausen’s disease): Clinicopathological spectrum with pathogenetic considerations. Int. J. Clin. Exp. Pathol. 2012, 5, 852–862. [Google Scholar]
- Santoro, C.; Perrotta, S.; Picariello, S.; Sicilipoti, M.; Cirillo, M.; Quaglietta, L.; Cinalli, G.; Cioffi, D.; Di lorgi, N.; Maghnie, M.; et al. Pretreatment endocrine disorders due to optic pathway gliomas in pediatric neurofibromatosis type 1: Multicenter study. J. Clin. Endocrinol. Metab. 2020, 105, dgaa138. [Google Scholar] [CrossRef]
- Bergsland, E. Somatostatinoma: Clinical Manisfestations, Diagnosis, and Management. In Post; Nathan, D.M., Whitcomb, D.C., Grover, S., Eds.; UptoDate. Available online: https://www.uptodate.com/contents/somatostatinoma-clinical-manifestations-diagnosis-and-management (accessed on 19 September 2019).
- Garbrecht, N.; Anlauf, M.; Schmitt, A.; Henopp, T.; Sipos, B.; Raffel, A.; Eisenberger, C.F.; Knoefel, W.T.; Pavel, M.; Fottner, C.; et al. Somatostatin-producing neuroendocrine tumors of the duodenum and pancreas: Incidence, types, biological behavior, association with inherited syndromes, and functional activity. Endocrine-Relat. Cancer 2008, 15, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Yamasaki, S.; Matsushita, H.; Ozawa, Y.; Kurosaki, A.; Takeuchi, K.; Hoshihara, Y.; Doi, T.; Watanabe, G.; Kawaminami, K. Duodenal somatostatinoma: A case report and review of 31 cases with special reference to the relationship between tumor size and metastasis. Pathol. Int. 2000, 50, 146–152. [Google Scholar] [CrossRef]
- Deppen, S.A.; Liu, E.; Blume, J.D.; Clanton, J.; Shi, C.; Jones-Jackson, L.B.; Lakhani, V.; Baum, R.P.; Berlin, J.; Smith, G.T.; et al. Safety and efficacy of 68Ga-DOTATATE PET/CT for diagnosis, staging, and treatment management of neuroendocrine tumors. J. Nucl. Med. 2016, 57, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Sadowski, S.M.; Neychev, V.; Millo, C.; Shih, J.; Nilubol, N.; Herscovitch, P.; Pacak, K.; Marx, S.J.; Kebebew, E. Prospective study of 68Ga-DOTATATE positron emission tomography/computed tomography for detecting gastro-entero-pancreatic neuroendocrine tumors and unknown primary sites. J. Clin. Oncol. 2016, 34, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fendrich, V.; Langer, P.; Celik, I.; Bartsch, D.K.; Zielke, A.; Ramaswamy, A.; Rothmund, M. An aggressive surgical approach leads to long-term survival in patients with pancreatic endocrine tumors. Ann. Surg. 2006, 244, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.; Saltz, L. Islet cell tumors of the pancreas: The medical oncologist’s perspective. Surg. Clin. North. Am. 2001, 81, 527–542. [Google Scholar] [CrossRef]
- King, J.; Quinn, R.; Glenn, D.M.; Janssen, J.; Tong, D.; Liaw, W.; Morris, D.L. Radioembolization with selective internal radiation microspheres for neuroendocrine liver metastases. Cancer 2008, 113, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Vinik, A.; Moattari, A.R. Use of somatostatin analog in management of carcinoid syndrome. Dig. Dis. Sci. 1989, 34 (Suppl. S3), 14S–27S. [Google Scholar] [CrossRef] [Green Version]
- Oberg, K.; Eriksson, B.; Lundqvist, M. Neuroendocrine tumours of the upper gastrointestinal tract and pancreas. Acta Chir. Scand. Suppl. 1988, 541, 76–85. [Google Scholar]
- Angeletti, S.; Corleto, V.D.; Schillaci, O.; Marignani, M.; Annibale, B.; Moretti, A.; Silecchia, G.; Scopinaro, F.; Basso, N.; Bordi, C.; et al. Use of the somatostatin analogue octreotide to localise and manage somatostatin-producing tumours. Gut 1998, 42, 792–794. [Google Scholar] [CrossRef] [Green Version]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef]
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988, 45, 575–578.
- Edge, S.B.; Compton, C.C.; Fritz, A.G. The American Joint Committee on Cancer: The 7th Edition of the AJCC Cancer Staging Manual and the Future of TNM; Greene, F.L., Trotti, A., Eds.; Springer: New York, NY, USA, 2010; ISBN 978-0-387-88442-4. [Google Scholar]
- National Comprehensive Cancer Network. Neuroendocrine and Adrenal Tumors (Version 1.2019); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2019. [Google Scholar]
- Kumar, T.; Gupta, B.; Das, P.; Jain, D.; Jain, H.A.; Madhusudhan, K.S.; Dash, N.R.; Gupta, S.D. Combined presence of multiple gastrointestinal stromal tumors along with duodenal submucosal somatostatinoma in a patient with neurofibromatosis type 1. Indian J. Pathol. Microbiol. 2016, 59, 359–361. [Google Scholar] [CrossRef]
- Lodish, M.B.; Stratakis, C.A. Endocrine tumours in neurofibromatosis type 1, tuberous sclerosis and related syndromes. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferner, R.E. Neurofibromatosis 1 and neurofibromatosis 2: A twenty first century perspective. Lancet Neurol. 2007, 6, 340–351. [Google Scholar] [CrossRef]
- Stewart, D.R.; Korf, B.R.; Nathanson, K.L.; Stevenson, D.A.; Yohay, K. Care of adults with neurofibromatosis type 1: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantor, A.M.; Rigby, C.C.; Beck, P.R.; Mangion, D. Neurofibromatosis, phaeochromocytoma, and somatostatinoma. BMJ 1982, 285, 1618–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappelli, C.; Agosti, B.; Braga, M.; Cumetti, D.; Gandossi, E.; Rizzoni, D.; Agabiti Rosei, E. Von Recklinghausen’s neurofibromatosis associated with duodenal somatostatinoma. A case report and review of the literature. Minerva Endocrinol. 2004, 29, 19–24. [Google Scholar]
- Mao, C.; Shah, A.; Hanson, D.J.; Howard, J.M. Von Recklinghausen’s disease associated with duodenal somatostatinoma: Contrast of duodenal versus pancreatic somatostatinomas. J. Surg. Oncol. 1995, 59, 67–73. [Google Scholar] [CrossRef]
- Relles, D.; Baek, J.; Witkiewicz, A.; Yeo, C.J. Periampullary and duodenal neoplasms in neurofibromatosis type 1: Two cases and an updated 20-year review of the literature yielding 76 cases. J. Gastrointest. Surg. 2010, 14, 1052–1061. [Google Scholar] [CrossRef]
- Adams, M.S.; Bronner-Fraser, M. Review: The role of neural crest cells in the endocrine system. Endocr. Pathol. 2009, 20, 92–100. [Google Scholar] [CrossRef]
- Kim, J.A.; Choi, W.H.; Kim, C.N.; Moon, Y.S.; Chang, S.H.; Lee, H.R. Duodenal somatostatinoma: A case report and review. [published correction appears in Korean J. Intern. Med. 26 June 2011, 253]. Korean J. Intern. Med. 2011, 26, 103–107. [Google Scholar] [CrossRef]
- Al-Risi, E.S.; Al-Essry, F.S.; Mula-Abed, W.S. Chromogranin A as a biochemical marker for neuroendocrine tumors: A single center experience at Royal Hospital, Oman. Oman Med. J. 2017, 32, 365–370. [Google Scholar] [CrossRef]
- Gut, P.; Czarnywojtek, A.; Fischbach, J.; Bączyk, M.; Ziemnicka, K.; Wrotkowska, E.; Gryczyńska, M.; Ruchała, M. Chromogranin A—Unspecific neuroendocrine marker. Clinical utility and potential diagnostic pitfalls. Arch. Med. Sci. 2016, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dluhy, R.G. Pheochromocytoma—Death of an axiom. N. Engl. J. Med. 2002, 346, 1486–1488. [Google Scholar] [CrossRef] [PubMed]
- Srirajaskanthan, R.; Kayani, I.; Quigley, A.M.; Soh, J.; Caplin, M.E.; Bomanji, J. The role of 68Ga-DOTATATE PET in patients with neuroendocrine tumors and negative or equivocal findings on 111In-DTPA-octreotide scintigraphy. J. Nucl. Med. 2010, 51, 875–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenning, E.P.; Kwekkeboom, D.J.; Bakker, W.H.; Breeman, W.A.; Kooij, P.P.; Oei, H.Y.; van Hagen, M.; Postema, P.T.; de Jong, M.; Reubi, J.C. Somatostatin receptor scintigraphy with [111In-DTPA-D-Phe1]- and [123I-Tyr3]-octreotide: The Rotterdam experience with more than 1000 patients. Eur. J. Nucl. Med. 1993, 20, 716–731. [Google Scholar] [CrossRef] [Green Version]
- Asnacios, A.; Courbon, F.; Rochaix, P.; Bauvin, E.; Cances-Lauwers, V.; Susini, C.; Schulz, S.; Boneu, A.; Guimbaud, R.; Buscail, L. Indium-111-pentetreotide scintigraphy and somatostatin receptor subtype 2 expression: New prognostic factors for malignant well-differentiated endocrine tumors. J. Clin. Oncol. 2008, 26, 963–970. [Google Scholar] [CrossRef]
- Wong, K.K.; Cahill, J.M.; Frey, K.A.; Avram, A.M. Incremental value of 111-in pentetreotide SPECT/CT fusion imaging of neuroendocrine tumors. Acad. Radiol. 2010, 17, 291–297. [Google Scholar] [CrossRef]
- Rinke, A.; Wittenberg, M.; Schade-Brittinger, C.; Aminossadati, B.; Ronicke, E.; Gress, T.M.; Müller, H.H.; Arnold, R.; PROMID Study Group. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors (PROMID): Results of long-term survival. Neuroendocrinology 2017, 104, 26–32. [Google Scholar] [CrossRef]
- Qian, Z.R.; Li, T.; Ter-Minassian, M.; Yang, J.; Chan, J.A.; Brais, L.K.; Masugi, Y.; Thiaglingam, A.; Brooks, N.; Nishihara, R.; et al. Association between somatostatin receptor expression and clinical outcomes in neuroendocrine tumors. Pancreas 2016, 45, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Vezzosi, D.; Bennet, A.; Rochaix, P.; Courbon, F.; Selves, J.; Pradere, B.; Buscail, L.; Susini, C.; Caron, P. Octreotide in insulinoma patients: Efficacy on hypoglycemia, relationships with Octreoscan scintigraphy and immunostaining with anti-sst2A and anti-sst5 antibodies. Eur. J. Endocrinol. 2005, 152, 757–767. [Google Scholar] [CrossRef]
- Baldelli, R.; Barnabei, A.; Rizza, L.; Isidori, A.M.; Rota, F.; Di Giacinto, P.; Paoloni, A.; Torino, F.; Corsello, S.M.; Lenzi, A.; et al. Somatostatin analogs therapy in gastroenteropancreatic neuroendocrine tumors: Current aspects and new perspectives. Front. Endocrinol. 2014, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Pavel, M.; Öberg, K.; Falconi, M.; Krenning, E.P.; Sundin, A.; Perren, A.; Berruti, A.; ESMO Guidelines Committee. Gastroenteropancreatic neuroendocrine neoplasms: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 844–860. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Elliott, E.; Barriuso, J.; Backen, A.; McNamara, M.G.; Hubner, R.; Valle, J.W. Chemotherapy for advanced non-pancreatic well-differentiated neuroendocrine tumours of the gastrointestinal tract, a systematic review and meta-analysis: A lost cause? Cancer Treat. Rev. 2016, 44, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Jefford, M.; Lai-Kwon, J.; Thai, A.; Hicks, R.J.; Michael, M. CAPTEM in Metastatic well-differentiated intermediate to high grade neuroendocrine tumors: A single centre experience. J. Oncol. 2019, 2019, Article. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, K.; Voros, B.A.; Meadows-Taylor, M.; Smeltzer, M.P.; Griffin, R.; Boudreaux, J.P.; Thiagarajan, R.; Woltering, E.A.; Ramirez, R.A. Outcomes of Capecitabine and Temozolomide (CAPTEM) in advanced Neuroendocrine Neoplasms (NENs). Cancers 2020, 12, 206. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.H.; Alexander, A.J.; Konda, B.; Wei, L.; Hemminger, J.A.; Schmidt, C.R.; Abdel-Misih, S.; Dillhoff, M.E.; Sipos, J.A.; Kirschner, L.S.; et al. Combination therapy with capecitabine and temozolomide in patients with low and high grade neuroendocrine tumors, with an exploratory analysis of O6-methylguanine DNA methyltransferase as a biomarker for response. Oncotarget 2017, 8, 104046–104056. [Google Scholar] [CrossRef]
- Walter, T.; van Brakel, B.; Vercherat, C.; Hervieu, V.; Forestier, J.; Chayvialle, J.A.; Molin, Y.; Lombard-Bohas, C.; Joly, M.O.; Scoazec, J.Y. O6-Methylguanine-DNA methyltransferase status in neuroendocrine tumours: Prognostic relevance and association with response to alkylating agents. Br. J. Cancer 2015, 112, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Carbonero, R.; Sorbye, H.; Baudin, E.; Raymond, E.; Wiedmann, B.; Niederle, B.; Sedlackova, E.; Toumpanakis, C.; Anlauf, M.; Cwifla, J.B.; et al. ENETS consensus guidelines for high-grade gastroenteropancreatic neuroendocrine tumors and neuroendocrine carcinomas. Neuroendocrinology 2016, 103, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, R.D.; Noonan, K.L.; Pavlovich, P.; Kennecke, H.F.; Lim, H.J. Outcomes of patients treated with capecitabine and temozolamide for advanced pancreatic neuroendocrine tumors (PNETs) and non-PNETs. J. Gastrointest. Oncol. 2014, 5, 247–252. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Fine, R.L.; Choi, J.; Nasir, A.; Coppola, D.; Chen, D.T.; Helm, J.; Kvols, L. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. C Cancer 2011, 117, 268–275. [Google Scholar] [CrossRef]
- Kouvaraki, M.A.; Ajani, J.A.; Hoff, P.; Wolff, R.; Evans, D.B.; Lozano, R.; Yao, J.C. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. [published correction appears in J. Clin. Oncol. 1 January 2005, 23, 248]. J. Clin. Oncol. 2004, 22, 4762–4771. [Google Scholar] [CrossRef]
- Fazio, N.; Buzzoni, R.; Delle Fave, G.; Tesselaar, M.E.; Wolin, E.; Van Cutsem, E.; Tomassetti, P.; Strosberg, J.; Voi, M.; Bubuteishvili-Pacaud, L.; et al. Everolimus in advanced, progressive, well-differentiated, non-functional neuroendocrine tumors: RADIANT-4 lung subgroup analysis. Cancer Sci. 2018, 109, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, R.J.; Kwekkeboom, D.J.; Krenning, E.; Bodei, L.; Grozinsky-Glasberg, S.; Arnold, R.; Borbath, I.; Cwikla, J.; Toumpanakis, C.; Kaltsas, G.; et al. ENETS consensus guidelines for the standards of care in neuroendocrine neoplasia: Peptide receptor radionuclide therapy with radiolabeled somatostatin analogues. Neuroendocrinology 2017, 105, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. NETTER-1 trial investigators. Phase 3 trial of 177Lu-dotatate for midgut neuroendocrine tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Claringbold, P.G.; Price, R.A.; Turner, J.H. Phase I-II study of radiopeptide 177Lu-octreotate in combination with capecitabine and temozolomide in advanced low-grade neuroendocrine tumors. Cancer Biother. Radiopharm. 2012, 27, 561–569. [Google Scholar] [CrossRef] [PubMed]













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, S.; Fica, S.; Parfeni, O.; Popa, L.; Manuc, T.; Rizea, O.; Lupescu, I.; Gherghe, M.; Becheanu, G.; Croitoru, A. Somatostatinoma and Neurofibromatosis Type 1-A Case Report and Review of the Literature. Diagnostics 2020, 10, 620. https://doi.org/10.3390/diagnostics10090620
Martin S, Fica S, Parfeni O, Popa L, Manuc T, Rizea O, Lupescu I, Gherghe M, Becheanu G, Croitoru A. Somatostatinoma and Neurofibromatosis Type 1-A Case Report and Review of the Literature. Diagnostics. 2020; 10(9):620. https://doi.org/10.3390/diagnostics10090620
Chicago/Turabian StyleMartin, Sorina, Simona Fica, Ovidiu Parfeni, Liliana Popa, Teodora Manuc, Oana Rizea, Ioana Lupescu, Mirela Gherghe, Gabriel Becheanu, and Adina Croitoru. 2020. "Somatostatinoma and Neurofibromatosis Type 1-A Case Report and Review of the Literature" Diagnostics 10, no. 9: 620. https://doi.org/10.3390/diagnostics10090620